mkdir -p monkeyflower
cd monkeyflowerVariant filtering
Compute environment setup
If you haven’t already done so, please read Compute environment for information on how to prepare your working directory.
In this exercise we will look at ways of filtering variant data. We will begin by applying filters to the variant file containing variant sites only, followed by an approach that filters on sequencing depth in a variant file containing both variant and invariant sites. The latter methodology can then be generalized to generate depth-based filters from BAM files.
Important
The commands of this document have been run on a subset (a subregion) of the data. Therefore, although you will use the same commands, your results will differ from those presented here.
Learning objectives
- filter variants by quality, read depth, and other metrics
- apply filters to a VCF file
- create per sample, per population, and total depth of coverage profiles
- generate mask files for downstream processing
Data setup
Move to your course working directory /proj/naiss2023-22-1084/users/USERNAME, create an exercise directory called monkeyflower and cd to it:
cd /proj/naiss2023-22-1084/users/USERNAME
mkdir -p monkeyflower
cd monkeyflowerRetrieve the data with rsync. You can use the --dry-run option to see what is going to happen; just remove it when you are content:
rsync --dry-run -av \
/proj/naiss2023-22-1084/webexport/monkeyflower/LG4/* .
# Remove the dry run option to actually retrieve data. The dot is
# important!
rsync -av /proj/naiss2023-22-1084/webexport/monkeyflower/LG4/* .Create an exercise directory called monkeyflower and cd to it:
Retrieve the variant files from https://export.uppmax.uu.se/naiss2023-22-1084/monkeyflower/LG4 with wget1:
wget --user pgip --password PASSWORD \
--recursive --accept='*.*' \
--reject='*.gif','index*' \
--no-parent --no-directories \
--no-clobber \
--directory-prefix=. \
https://export.uppmax.uu.se/naiss2023-22-1084/monkeyflower/LG4/
Tools
Execute the following command to load modules:
module load uppmax bioinfo-tools \
mosdepth/0.3.3 BEDTools/2.29.2 \
samtools/1.17 bcftools/1.17 \
vcftools/0.1.16 vcflib/1.0.1csvtk has been manually added to the module system and can be loaded as follows:
module use /proj/naiss2023-22-1084/modules
module load csvtkCopy the contents to a file environment.yml and install packages with mamba env update -f environment.yml.
channels:
- conda-forge
- bioconda
- default
dependencies:
- bedtools=2.31.0
- bcftools=1.15.1
- csvtk=0.28.0
- mosdepth=0.3.3
- samtools=1.15.1
- vcflib=1.0.1
- vcftools=0.1.16Background
Regardless of how a raw variant call set has been produced, the calls will be of varying quality for a number of reasons. For high-coverage sequencing, the two most common are incompleteness of the reference sequence and misalignments in repetitive regions (Li, 2014). Low-coverage sequencing comes with its own biases and issues, with the most important being the difficulty to accurately call genotypes (Maruki & Lynch, 2017).
In order to improve the accuracy of downstream inference, a number of analysis-dependent quality control filters should be applied to the raw variant call set (for a concise summary, see Lou et al. (2021)). In this exercise, we will begin by applying filters to the variant file containing variant sites only, followed by a more general approach based on depth filtering of a variant file consisting of all sites, variant as well as invariant. If time permits, we will also look at an approach that generates depth profiles directly from read mappings (BAM files) and that works also when it is impractical due to file size to include invariant sites in the variant file.
It is worthwhile to spend time thinking about filtering. As we will see, there are numerous metrics to filter on, and different applications require different filters. This is not as straightforward as it first may seem, and even experts struggle to get filtering settings right.
Some recommended data filters
There are many ways to filter data. Choosing the right set of filters is not easy, and choosing appropriate thresholds depends on application, among other things. Below we list some recommended data filters and thresholds that have general applicability and recently have been reviewed (Lou et al., 2021, Table 3):
- depth: Given the difficulty of accurately genotyping low-coverage sites, it is recommended to set a minimum read depth cutoff to remove false positive calls. It is also recommended to set a maximum depth cutoff as excessive coverage is often due to mappings to repetitive regions. The thresholds will depend on the depth profile over all sites, but is usually chosen as a range around the mean or median depth (e.g. lower threshold 0.8X mean, upper threshold median + 2 standard deviations).
- minimum number of individuals: To avoid sites with too much missing data across individuals, a common requirement is that a minimum number (fraction) of individuals, say 75%, have sequence coverage (depth-based filter) or genotype calls.
- quality (p-value): Most variant calling software provide a Phred-scaled probability score that a genotype is a true genotype. Quality values below 20 (i.e., 1%) should not be trusted, but could be set much higher (i.e., lower p-value) depending on application. Note that if a VCF file includes invariant sites, they have quality values set to 0, which renders quality based filtering inappropriate.
- MAF: Filter sites based on a minimum minor allele frequency (MAF) threshold. The appropriate choice depends on application. For instance, for PCA or admixture analyses, low-frequency SNPs are uninformative, and a reasonably large cutoff (say, 0.05-0.10) could be set. If an analysis depends on invariant sites, this filter should not be applied.
Important
For applications where invariant sites should be included, such as genetic diversity calculations, neither quality nor MAF filtering should be applied.
Basic filtering of a VCF file2
We will begin by creating filters for a VCF file consisting of variant sites only for red and yellow ecotypes. Before we start, let’s review some statistics for the entire (unfiltered) call set:
bcftools stats variantsites.vcf.gz | grep ^SNSN 0 number of samples: 10
SN 0 number of records: 128395
SN 0 number of no-ALTs: 0
SN 0 number of SNPs: 105508
SN 0 number of MNPs: 0
SN 0 number of indels: 23086
SN 0 number of others: 0
SN 0 number of multiallelic sites: 10412
SN 0 number of multiallelic SNP sites: 2108Keep track of these numbers as we will use them to evaluate the effects of filtering.
Generate random subset of variants
Depending on the size of a study, both in terms of reference sequence and number of samples, the VCF output can become large; a VCF file may in fact contain millions of variants and be several hundred GB! We want to create filters by examining distributions of VCF quality metrics and setting reasonable cutoffs. In order to decrease run time, we will look at a random sample of sites. We use the vcflib program vcfrandomsample to randomly sample approximately 100,000 sites from our VCF file3:
# Set parameter r = 100000 / total number of variants
bcftools view variantsites.vcf.gz | vcfrandomsample -r 0.8 |\
bgzip -c > variantsites.subset.vcf.gz
bcftools index variantsites.subset.vcf.gz
bcftools stats variantsites.subset.vcf.gz |\
grep "number of records:"SN 0 number of records: 103044The -r parameter sets the rate of sampling which is why we get approximately 100,000 sites. You will need to adjust this parameter accordingly.
We will now use vcftools to compile statistics. By default, vcftools outputs results to files with a prefix out. in the current directory. You can read up on settings and options by consulting the man pages with man vcftools4. Therefore, we define a variable OUT where we will output our quality metrics, along with a variable referencing our variant subset:
mkdir -p vcftools
OUT=vcftools/variantsites.subset
VCF=variantsites.subset.vcf.gzGenerate statistics for filters
vcftools can compile many different kinds of statistics. Below we will focus on the ones relevant to our data filters. We will generate metrics and plot results as we go along, with the goal of generating a set of filtering thresholds to apply to the data.
Total depth per site
To get a general overview of depth of coverage, we first generate the average depth per sample5:
vcftools --gzvcf $VCF --depth --out $OUT 2>/dev/null
cat ${OUT}.idepth
csvtk summary -t -f MEAN_DEPTH:mean ${OUT}.idepthINDV N_SITES MEAN_DEPTH
PUN-R-ELF 102847 8.56957
PUN-R-JMC 102833 9.54936
PUN-R-LH 102876 9.24544
PUN-R-MT 102826 9.44265
PUN-R-UCSD 102917 8.94361
PUN-Y-BCRD 102839 9.92774
PUN-Y-INJ 102808 8.26867
PUN-Y-LO 102807 7.82653
PUN-Y-PCT 102798 9.18246
PUN-Y-POTR 102774 8.50441
MEAN_DEPTH:mean
8.95The average coverage over all samples is 8.9X. This actually is in the low range for a protocol based on explicitly calling genotypes. At 5X coverage, there may be a high probability that only one of the alleles has been sampled (Nielsen et al., 2011), whereby sequencing errors may be mistaken for true variation.
Then we calculate depth per site to see if we can identify reasonable depth cutoffs:
vcftools --gzvcf $VCF --site-depth --out $OUT 2>/dev/null
head -n 3 ${OUT}.ldepthCHROM POS SUM_DEPTH SUMSQ_DEPTH
LG4 2130 11 21
LG4 2139 12 26So, for each position, we have a value (column SUM_DEPTH) for the total depth across all samples.
We plot the distribution of total depths by counting how many times each depth is observed. This can be done with csvtk summary where we count positions and group (-g) by the SUM_DEPTH value:6
csvtk summary -t -g SUM_DEPTH -f POS:count -w 0 ${OUT}.ldepth |\
csvtk sort -t -k 1:n |\
csvtk plot line -t - -x SUM_DEPTH -y POS:count \
--point-size 0.01 --xlab "Depth of coverage (X)" \
--ylab "Genome coverage (bp)" \
--width 9.0 --height 3.5 > $OUT.ldepth.png
On csvtk as plotting software
You are of course perfectly welcome to use R or some other software to make these plots. We choose to generate the plots using csvtk to avoid too much context switching, and also because it emulates much of the functionality in R, albeit much less powerful when it comes to plotting.
As Figure 1 shows, most sites fall within a peak, but also that there are sites with very high coverage, up to ten times as high as the depth at the peak maximum. We calculate some range statistics to get an idea of the spread. The following csvtk command will calculate the minimum, first quartile, median, mean, third quartile, and maximum of the third column (SUM_DEPTH):
csvtk summary -t -f 3:min,3:q1,3:median,3:mean,3:q3,3:max,3:stdev vcftools/variantsites.subset.ldepthSUM_DEPTH:min SUM_DEPTH:q1 SUM_DEPTH:median SUM_DEPTH:mean SUM_DEPTH:q3 SUM_DEPTH:max SUM_DEPTH:stdev
1.00 55.00 72.00 89.28 88.00 33988.00 245.06The range from the first quartile (q1) to the third (q3) is 55-88, showing most sites have a depth between 50-100X. We redraw the plot to zoom in on the peak:

--x-min 0 --x-max 140 --y-max 2500 to the plotting call.We could choose a filter based on the quantile statistics above, or by eye-balling the graph. In this example, we could have chosen the range 50-150X, which equates to 5-15X depth per sample; note that your values will probably be different.
As an aside, we mention that there is a command to directly get the per-site mean depth, --site-mean-depth:
vcftools --gzvcf $VCF --site-mean-depth --out $OUT 2>/dev/null
head -n 3 ${OUT}.ldepth.meanCHROM POS MEAN_DEPTH VAR_DEPTH
LG4 2130 1.1 0.988889
LG4 2139 1.2 1.28889Variant quality distribution
Another quantity of interest is the variant quality. Recall, variant quality scores are Phred-scaled scores, meaning a value of 10 has a 10% chance of being wrong, 20 a 1% chance, and so on; you typically want to at least filter on 20 or even higher. We extract and plot the quality values below:
vcftools --gzvcf $VCF --site-quality --out $OUT# To improve histogram, filter out extreme quality scores. You
# may have to fiddle with the exact values
csvtk filter -t -f "QUAL>0" -f "QUAL<1000" ${OUT}.lqual | \
csvtk summary -t -g QUAL -f POS:count -w 0 - |\
csvtk sort -t -k 1:n |\
csvtk plot hist -t --bins 100 - \
--xlab "Quality value" \
--ylab "Count" \
--width 9.0 --height 3.5 > $OUT.lqual.png
Clearly most quality scores are above 20-30. For many applications, we recommend setting 30 as the cutoff.
Minor allele frequency distribution
Since we are going to calculate nucleotide diversities, we will not filter on the minor allele frequency (MAF) here. Nevertheless, we generate the distribution and plot for discussion purposes. The --freq2 will output the frequencies only, adding the option --max-alleles 2 to focus only on bi-allelic sites:
vcftools --gzvcf $VCF --freq2 --out $OUT --max-alleles 2 2>/dev/null
head -n 3 ${OUT}.frqCHROM POS N_ALLELES N_CHR {FREQ}
LG4 2130 2 14 0.857143 0.142857
LG4 2139 2 8 0.25 0.75The last two columns are frequencies ordered according to the reference allele. Therefore, we need to pick the minimum value to get the MAF. We can use csvtk mutate to create a new column
csvtk fix -t ${OUT}.frq 2>/dev/null |\
csvtk mutate2 -t -n maf -e '${5} > ${6} ? "${6}" : "${5}" ' - |\
csvtk plot hist -t --bins 20 -f maf - \
--xlab "Minor allele frequency" \
--ylab "Count" \
--width 9.0 --height 3.5 > $OUT.frq.png
Since our variant file consists of 10 individuals, that is, 20 chromosomes, there are only so many frequencies that we can observe, which is why the histogram looks a bit disconnected7. In fact, given 20 chromosomes, MAF=0.05 corresponds to one alternative allele among all individuals (singleton), MAF=0.1 to two, and so on. The maximum value is 0.5, which is to be expected, as it is a minor allele frequency. We note that there are more sites with a low minor allele frequency, which in practice means there are many singleton variants.
This is where filtering on MAF can get tricky. Singletons may correspond to sequencing error, but if too hard a filter is applied, the resulting site frequency spectrum (SFS) will be skewed. For statistics that are based on the SFS, this may lead biased estimates. Since we will be applying such a statistic, we do not filter on the MAF here. Note, however, that for other applications, such as population structure, it may be warranted to more stringently (say, MAF>0.1) filter out low-frequency variants.
Missing data for individuals and sites
The proportion missing data per individual can indicate whether the input DNA was of poor quality, and that the individual should be excluded from analysis. Note that in this case, missing data refers to a missing genotype call and not sequencing depth!
We can calculate the proportion of missing data
vcftools --gzvcf $VCF --missing-indv --out $OUT 2>/dev/null
head -n 3 ${OUT}.imissINDV N_DATA N_GENOTYPES_FILTERED N_MISS F_MISS
PUN-R-ELF 103044 0 6991 0.0678448
PUN-R-JMC 103044 0 6588 0.0639339and look at the results, focusing on the F_MISS column (proportion missing sites):
csvtk plot hist -t --x-min 0 -f F_MISS ${OUT}.imiss > ${OUT}.imiss.png
Here, the proportion lies in the range 0.06-0.10 for all samples, which indicates good coverage of all samples and we refrain from taking any action.
Similarly, we can look at missingness per site. This is related to the filter based on minimum number of individuals suggested by Lou et al. (2021). We calculate
vcftools --gzvcf $VCF --missing-site --out $OUT 2>/dev/null
head -n 3 ${OUT}.lmissCHR POS N_DATA N_GENOTYPE_FILTERED N_MISS F_MISS
LG4 2130 20 0 6 0.3
LG4 2139 20 0 12 0.6and plot to get an idea of if there are sites that lack data.
csvtk plot hist --bins 20 -t -f F_MISS ${OUT}.lmiss > ${OUT}.lmiss.png
As Figure 6 shows, many sites have no or little missing data, but given the low coverage, there is a non-negligible number of sites with higher missingness. We calculate range statistics to get a feeling for a good cutoff:
csvtk summary -t -f 6:min,6:q1,6:median,6:mean,6:q3,6:max vcftools/variantsites.subset.lmissF_MISS:min F_MISS:q1 F_MISS:median F_MISS:mean F_MISS:q3 F_MISS:max
0.00 0.00 0.00 0.08 0.10 0.90The mean missingness is 8%, so we can safely use 25% missingness as threshold. Typical values of tolerated missingness lie in the range 5-25%. Note that vcftools interprets this value as 1 - missingness, so it has to be inverted to 75% when filtering!
Heterozygosity
vcftools can calculate the heterozygosity per individual. More specifically, it estimates the inbreeding coefficient F for each individual.
vcftools --gzvcf $VCF --het --out $OUT 2>/dev/null
cat ${OUT}.hetINDV O(HOM) E(HOM) N_SITES F
PUN-R-ELF 72393 65584.9 87301 0.31351
PUN-R-JMC 68270 65824.3 87571 0.11246
PUN-R-LH 70890 66054.7 87882 0.22153
PUN-R-MT 69985 65776.7 87480 0.19390
PUN-R-UCSD 74700 65580.4 87550 0.41510
PUN-Y-BCRD 69345 64909.7 86275 0.20759
PUN-Y-INJ 71169 64112.6 85018 0.33754
PUN-Y-LO 72845 63349.3 84415 0.45077
PUN-Y-PCT 74452 64462.2 85937 0.46519
PUN-Y-POTR 71716 64520.3 85782 0.33844Here, F is a measure of how much the observed homozygotes O(HOM) differ from the expected (E(HOM); expected by chance under Hardy-Weinberg equilibrium), and may be negative. vcftools calculates F from the expression \(F=(O-E)/(N-E)\)8, which you can verify by substituting the variables in the output.
If F is positive (\(O(HOM) > E(HOM)\)), i.e., there are more observed homozygotes than expected, then there is a deficit of heterozygotes, which could be a sign of inbreeding or signs of allelic dropout in case of low sequencing coverage.
If F is negative, there are fewer observed homozygotes than expected, or conversely, an excess of heterozygotes. This could be indicative of poor sequence quality (bad mappings) or contamination (Purcell et al., 2007).
The underlying assumption is HWE, which holds for F=0.
In this case, we know that the samples are from two different populations, red and yellow. In such cases, we actually expect a deficit of heterozygotes (and consequently, positive F) simply due to something called the Wahlund effect.
Warning
The inbreeding coefficient is a population-level statistic and is not reliable for small sample sizes (\(n<10\), say). Therefore, our sample size is in the lower range and the results should be taken with a grain of salt.
Filtering the VCF
Now that we have decided on filters we can apply them to the VCF. We first set the filters as variables:
MISS=0.75
QUAL=30
MIN_DEPTH=5
MAX_DEPTH=15and run vcftools as follows:
OUTVCF=${VCF%.subset.vcf.gz}.filtered.vcf.gz
vcftools --gzvcf $VCF \
--remove-indels --max-missing $MISS \
--min-meanDP $MIN_DEPTH --max-meanDP $MAX_DEPTH \
--minDP $MIN_DEPTH --maxDP $MAX_DEPTH --recode \
--stdout 2>/dev/null |
gzip -c > $OUTVCFCompare the results with the original input:
bcftools stats $OUTVCF | grep "^SN"SN 0 number of samples: 10
SN 0 number of records: 37697
SN 0 number of no-ALTs: 0
SN 0 number of SNPs: 37486
SN 0 number of MNPs: 0
SN 0 number of indels: 0
SN 0 number of others: 0
SN 0 number of multiallelic sites: 1666
SN 0 number of multiallelic SNP sites: 688Quite a substantial portion variants have in fact been removed, which here can most likely be attributed to the low average sequencing coverage.
Depth filtering of VCF with invariant sites
Now we turn our attention to a VCF file containing variant and invariant sites. We will generate depth-based filters, with the motivation that they represent portions of the genome that are accessible to analysis, regardless of whether they contain variants or not. In so doing, we treat filtered sites as missing data and do not assume that they are invariant, as many software packages do.

Figure 7 illustrates the sequencing coverage of three samples. The important thing to note is that the coverage is uneven. Some regions lack coverage entirely, e.g., due to random sampling or errors in the reference sequence. Other regions have excessive coverage, which could be a sign of repeats that have been collapsed in the reference. A general coverage filter could then seek to mask out sites where a fraction (50%, say) of individuals have too low or excessive coverage.
The right panel illustrates the sum of coverages across all samples. Minimum and maximum depth filters could be applied to the aggregate coverages of all samples, or samples grouped by population, to eliminate sites confounding data support.
As mentioned, the VCF in this exercise contains all sites; that is, both monomorphic and polymorphic sites are present. Every site contains information about depth and other metadata, which makes it possible to apply coverage filters directly to the variant file itself.
However, it may not always be possible to generate a VCF with all sites. Species with large genomes will produce files so large that they prevent efficient downstream processing. Under these circumstances, ad hoc coverage filters can be applied to the BAM files to in turn generate sequence masks that can be used in conjunction with the variant file. This is the topic for the advanced session (Section 4).
Regardless of approach, the end result is a set of regions that are discarded (masked) for a given analysis. They can be given either as a BED file, or a sequence mask, which is a FASTA-like file consisting of integer digits (between 0 and 9) for each position on a chromosome. Then, by setting a cutoff, an application can mask positions higher than that cutoff. We will generate mask files with 0 and 1 digits, an example of which is shown below, where the central 10 bases of the reference (top) are masked (bottom).
>LG4 LG4:12000001-12100000
GGACAATTACCCCCTCCGTTATGTTTCAGT
>LG4
000000000011111111110000000000Data summary and subset
We start by summarising the raw data, as before.
bcftools stats allsites.vcf.gz | grep ^SNSN 0 number of samples: 10
SN 0 number of records: 100000
SN 0 number of no-ALTs: 91890
SN 0 number of SNPs: 3980
SN 0 number of MNPs: 0
SN 0 number of indels: 1197
SN 0 number of others: 0
SN 0 number of multiallelic sites: 541
SN 0 number of multiallelic SNP sites: 69Data for depth filters
By now you should be familiar with the vcftools commands to generate relevant data for filters. In particular, we used --site-depth to generate depth profiles over all sites, and --missing-site to generate missingness data, based on genotype presence/abscence, for every site. Use these same commands again to generate a set of depth filters.
Exercise
Use vcflib and vcftools to select a subset of variants from which to generate data. Use vcftools commands --site-depth and --missing-site as before to
- generate data
- (possibly) compute summary statistics with
csvtk - plot depth distributions
- select thresholds for depth-based and missingness filters
- filter the input VCF
Call the final output file allsites.filtered.vcf.gz and compare your output to the input file.
Answer
allsites subset
# Set parameter r = 100000 / total number of variants; input file
# here consists of 100000 entries. Adjust this parameter.
bcftools view allsites.vcf.gz | vcfrandomsample -r 1.0 |\
bgzip -c > allsites.subset.vcf.gz
bcftools index allsites.subset.vcf.gz
bcftools stats allsites.subset.vcf.gz |\
grep "^SN"SN 0 number of samples: 10
SN 0 number of records: 100000
SN 0 number of no-ALTs: 91890
SN 0 number of SNPs: 3980
SN 0 number of MNPs: 0
SN 0 number of indels: 1197
SN 0 number of others: 0
SN 0 number of multiallelic sites: 541
SN 0 number of multiallelic SNP sites: 69mkdir -p vcftools
OUT=vcftools/allsites.subset
VCF=allsites.subset.vcf.gzDepth per site
vcftools --gzvcf $VCF --site-depth --out $OUT 2>/dev/null
head -n 3 ${OUT}.ldepth
csvtk summary -t -f 3:min,3:q1,3:median,3:mean,3:q3,3:max,3:stdev ${OUT}.ldepthCHROM POS SUM_DEPTH SUMSQ_DEPTH
LG4 1 15 37
LG4 2 16 38
SUM_DEPTH:min SUM_DEPTH:q1 SUM_DEPTH:median SUM_DEPTH:mean SUM_DEPTH:q3 SUM_DEPTH:max SUM_DEPTH:stdev
0.00 58.00 76.00 78.93 91.00 2041.00 92.21
Missingness
vcftools --gzvcf $VCF --missing-site --out $OUT 2>/dev/null
head -n 3 ${OUT}.lmiss
csvtk summary -t -f 6:min,6:q1,6:median,6:mean,6:q3,6:max ${OUT}.lmissCHR POS N_DATA N_GENOTYPE_FILTERED N_MISS F_MISS
LG4 1 20 0 6 0.3
LG4 2 20 0 4 0.2
F_MISS:min F_MISS:q1 F_MISS:median F_MISS:mean F_MISS:q3 F_MISS:max
0.00 0.00 0.10 0.16 0.20 1.00csvtk plot hist --bins 20 -t -f F_MISS ${OUT}.lmiss > ${OUT}.lmiss.png
Filter input
Unless there is any strange bias that leads to a difference in coverage between variant and invariant sites, the final values should be similar to those before. We set filters and generate the output:
MISS=0.75
MIN_DEPTH=5
MAX_DEPTH=15OUTVCF=${VCF%.subset.vcf.gz}.filtered.vcf.gz
vcftools --gzvcf $VCF \
--remove-indels --max-missing $MISS \
--min-meanDP $MIN_DEPTH --max-meanDP $MAX_DEPTH \
--minDP $MIN_DEPTH --maxDP $MAX_DEPTH --recode \
--stdout 2>/dev/null |
gzip -c > $OUTVCFCompare the results with the original input:
bcftools stats $OUTVCF | grep "^SN"SN 0 number of samples: 10
SN 0 number of records: 46691
SN 0 number of no-ALTs: 43386
SN 0 number of SNPs: 2244
SN 0 number of MNPs: 0
SN 0 number of indels: 0
SN 0 number of others: 0
SN 0 number of multiallelic sites: 129
SN 0 number of multiallelic SNP sites: 43Genotype depth data and BED output
Instead of calculating missing genotypes per site, we can retrieve the individual depth for each genotype with --geno-depth. Since we know cutoffs for mean depth (5-15), we can run this command on the main input file (allsites.vcf.gz). For reasons that soon will become clear, we also rerun --site-depth-mean.
VCF=allsites.vcf.gz
OUT=vcftools/allsites
vcftools --gzvcf ${VCF} --geno-depth --out $OUT 2>/dev/null
vcftools --gzvcf ${VCF} --site-mean-depth --out $OUT 2>/dev/null
head -n 3 ${OUT}.gdepthCHROM POS PUN-R-ELF PUN-R-JMC PUN-R-LH PUN-R-MT PUN-R-UCSD PUN-Y-BCRD PUN-Y-INJ PUN-Y-LO PUN-Y-PCT PUN-Y-POTR
LG4 1 1 0 3 1 3 0 3 0 2 2
LG4 2 1 1 3 1 3 0 3 0 2 2We could then combine these two files and perform filtering as follows: for each site check that
- the mean depth is within the filter range
- there is a minimimum number of genotypes with sufficient depth
- individual genotype depth does not exceed a maximum depth threshold
If any of the points above fail, the site is discarded. We keep track of sites that pass the filters and output positions in BED format (a 0-based tab-separated format consisting of columns chrom, chromStart, and chromEnd).
Here is some code to achieve these goals. Unfortunately csvtk doesn’t seem to have support for calculating column margins out of the box, which is why we have to resort to this complicated construct using awk to count the number of individual genotypes that pass the coverage threshold 5.
BEDOUT=${VCF%.vcf.gz}.keep.bed
csvtk join -t ${OUT}.ldepth.mean ${OUT}.gdepth -f CHROM,POS |\
csvtk filter -t -f "MEAN_DEPTH>=5" |\
csvtk filter -t -f "MEAN_DEPTH<=15" |\
awk -v FS="\t" -v OFS="\t" \
'NR > 1 {count=0; for (i=4; i<=NF; i++)\
{if ($i>4) count++ }; if (count>=5) print $1, $2 - 1, $2}'|\
bedtools merge > ${BEDOUT}
head -n 3 $BEDOUTLG4 76 99
LG4 278 336
LG4 346 409The BED file contains a list of regions that are accessible to analysis.
Sequence masks
In addition to the BED output files, we can generate sequence masks. First, we set a variable to point to the reference sequence and index it.
export REF=M_aurantiacus_v1.fasta
samtools faidx ${REF}Now, we use the command bedtools makefasta to make a sequence mask file in FASTA format consisting solely of 1’s:9
awk 'BEGIN {OFS="\t"} {print $1, 0, $2}' ${REF}.fai > ${REF}.bed
bedtools maskfasta -fi ${REF} -mc 1 -fo ${REF}.mask.fa -bed ${REF}.bedWe generate a file where all positions are masked because the BED files contain regions that we want to keep. Therefore, we want to convert corresponding positions to zeros. This file will be used as a template for all mask files.
We then apply bedtools maskfasta again to unmask (set to 0) the positions that overlap with the BED coordinates:
bedtools maskfasta -fi ${REF}.mask.fa -mc 0 -fo ${REF}.unmask.fa \
-bed allsites.keep.bed
head -n 3 ${REF}.unmask.fa>LG4
111111111111111111111111111111111111111111111111111111111111
111111111111111100000000000000000000000111111111111111111111We can convince ourselves that this has worked by counting the number of unmasked positions in both the BED file (with bedtools genomecov) and sequence mask:
bedtools genomecov -i allsites.keep.bed -g ${REF}.fai | grep genome
# tr: -d deletes all characters not (-c, complement) in the character
# set '0'. wc: -m option counts characters
cat ${REF}.unmask.fa | tr -d -c '0' | wc -mgenome 0 20935 100000 0.20935
genome 1 79065 100000 0.79065
79065Note that 0 and 1 in the bedtools genomecov output refers to coverage (i.e., absence/presence) and not unmask/mask as in the mask FASTA file.
Example: Using a sequence mask with vcftools
The sequence mask file can be used with vcftools with the option --mask. Before we use, however, we need to convert the mask file to one sequence per line10. seqkit is a neat tool that allows us to do this without hassle. As an example, we then perform a genetic diversity calculation with and without mask file to highlight the difference:
WIDEMASK=${REF}.unmask.wide.fa
seqkit seq -w 0 ${REF}.unmask.fa > ${WIDEMASK}
vcftools --gzvcf allsites.vcf.gz --mask $WIDEMASK \
--site-pi --stdout 2>/dev/null |\
csvtk summary -t --ignore-non-numbers --decimal-width 4 \
--fields PI:count,PI:mean
vcftools --gzvcf allsites.vcf.gz --site-pi --stdout 2>/dev/null |\
csvtk summary -t --ignore-non-numbers --decimal-width 4 \
--fields PI:count,PI:meanPI:count PI:mean
79065 0.0183
PI:count PI:mean
100000 0.0208Clearly, filtering may have significant impact on the final outcome. You must choose your filters wisely!
Advanced depth filtering on BAM files
Important
This exercise is optional. It was created prior to the previous filtering exercises and may contain overlapping explanations and information. The procedure closely resembles that from the section on genotype depth data above, but depth profiles are now calculated from BAM files, which requires different tools.
In this section, we will restrict our attention to coverage-based filters, with the aim of generating sequence masks to denote regions of a reference sequence that contain sufficient information across individuals and populations. Furthermore, the masks will be applied in the context of genetic diversity calculations, in which case specific filters on polymorphic sites (e.g., p-value or minimum minor allele frequency (MAF)) should not be applied (all sites contain information).
Mapped reads provide information about how well a given genomic region has been represented during sequencing, and this information is usually summarized as the sequencing coverage. For any locus, this is equivalent to the number of reads mapping to that locus.
Sequencing coverage is typically not uniformly distributed over the reference. Reasons may vary but include uneven mapping coverage due to repeat regions, low coverage due to mis-assemblies, or coverage biases generated in the sequencing process. Importantly, both variable and monomorphic sites must be treated identically in the filtering process to eliminate biases between the two kinds of sites.
In this part, we will use mosdepth and bedtools to quickly generate depth of coverage profiles of mapped data. mosdepth is an ultra-fast command line tool for calculating coverage from a BAM file. By default, it generates a summary of the global distribution, and per-base coverage in bed.gz format. We will be using the per-base coverage for filtering.
Alternatively, mosdepth can also output results in a highly compressed format d4, which has been developed to handle the ever increasing size of resequencing projects. Files in d4 format can be processed with the d4-tools tool (Hou et al., 2021). For instance, d4tools view will display the coverage in bed format. We mention this in passing as it may be relevant when working with large genomes or sample sizes, but given the size of our sample data, we will be using bedtools from now on.
Per sample coverage
We start by calculating per-sample coverages with mosdepth. For downstream purposes, we need to save the size of the chromosomes we’re looking at, and for many applications, a fasta index file is sufficient.
export REF=M_aurantiacus_v1.fasta
samtools faidx ${REF}The syntax to generate coverage information for a BAM file is mosdepth <prefix> <input file>. Here, we add the -Q option to exclude reads with a mapping quality less than 20:
mosdepth -Q 20 PUN-Y-INJ PUN-Y-INJ.sort.dup.recal.bamThe per-base coverage output file will be named PUN-Y-INJ.per-base.bed.gz and can be viewed with bgzip:
bgzip -c -d PUN-Y-INJ.per-base.bed.gz | head -n 5LG4 0 9 3
LG4 9 13 4
LG4 13 53 5
LG4 53 56 6
LG4 56 62 7To get an idea of what the coverage looks like over the chromsome, we will make use of two versatile programs for dealing with genomic interval data and delimited text files. Both programs operate on streams, enabling the construction of powerful piped commands at the command line (see below).
The first is bedtools, which consists of a set of tools to perform genome arithmetic on bed-like file formats. The bed format is a tab-delimited format that at its simplest consists of the three columns chrom (the chromosome name), chromStart, and chromEnd, the start and end coordinates of a region.
The second is csvtk, a program that provides tools for dealing with tab- or comma-separated text files. Avid R users will see that many of the subcommands are similar to those in the tidyverse dplyr package.
We can use these programs in a one-liner to generate a simple coverage plot (Figure 10)11
bedtools intersect -a <(bedtools makewindows -g ${REF}.fai -w 1000) \
-b PUN-Y-INJ.per-base.bed.gz -wa -wb | \
bedtools groupby -i - -g 1,2,3 -c 7 -o mean | \
csvtk plot -t line -x 2 -y 4 --point-size 0.01 --xlab Position \
--ylab Coverage --width 9.0 --height 3.5 > fig-plot-coverage.png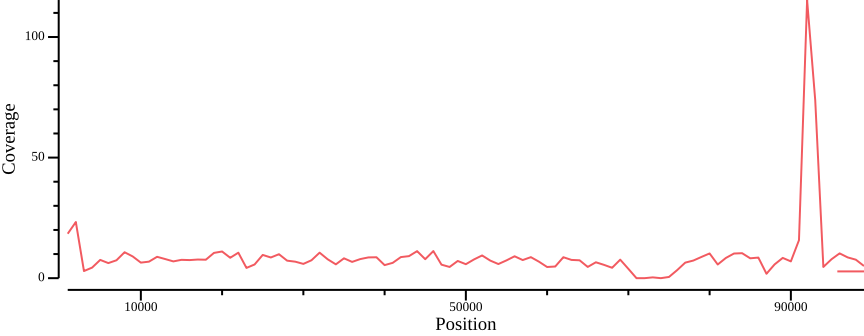
-w) parameter to change smoothing.Apparently there are some high-coverage regions that could be associated with, e.g., collapsed repeat regions in the assembly. Let’s compile coverage results for all samples, using bash string manipulation to generate file prefix12
# [A-Z] matches characters A-Z, * is a wildcard character that matches
# anything
for f in [A-Z]*.sort.dup.recal.bam; do
# Extract the prefix by removing .sort.dup.recal.bam
prefix=${f%.sort.dup.recal.bam}
mosdepth -Q 20 $prefix $f
# Print sample name
echo -e -n "$prefix\t"
# Get the summary line containing the total coverage
cat $prefix.mosdepth.summary.txt | grep total
done > ALL.mosdepth.summary.txt
# View the file
cat ALL.mosdepth.summary.txtPUN-R-ELF total 100000 845166 8.45 0 187
PUN-R-JMC total 100000 934032 9.34 0 244
PUN-R-LH total 100000 940246 9.40 0 225
PUN-R-MT total 100000 956272 9.56 0 233
PUN-R-UCSD total 100000 812507 8.13 0 293
PUN-Y-BCRD total 100000 882549 8.83 0 320
PUN-Y-INJ total 100000 818921 8.19 0 229
PUN-Y-LO total 100000 656112 6.56 0 151
PUN-Y-PCT total 100000 836694 8.37 0 179
PUN-Y-POTR total 100000 795201 7.95 0 205We can calculate the total coverage by summing the values of the fifth column with csvtk as follows:
csvtk summary -H -t ALL.mosdepth.summary.txt -f 5:sumto get the total coverage 84.78, which gives a hint at where the diploid coverage peak should be.
Sample set coverages
In this section, we will summarize coverage information for different sample sets, the motivation being that different filters may be warranted depending on what samples are being analysed. For instance, the coverage cutoffs for all samples will most likely be different from those applied to the subpopulations. In practice this means summing coverage tracks like those in Figure 10 for all samples.
We can combine the coverage output from different samples with bedtools unionbedg. We begin by generating a coverage file for all samples, where the output columns will correspond to individual samples. To save typing, we collect the sample names and generate matching BED file names to pass as arguments to options -names and -i, respectively. Also, we include positions with no coverage (-empty) which requires the use of a genome file (option -g). The BED output is piped to bgzip which compresses the output, before finally indexing with tabix13.
SAMPLES=$(csvtk grep -f Taxon -r -p "yellow" -r -p "red" sampleinfo.csv | csvtk cut -f SampleAlias | grep -v SampleAlias | tr "\n" " ")
BEDGZ=$(for sm in $SAMPLES; do echo -e -n "${sm}.per-base.bed.gz "; done)
bedtools unionbedg -header -names $SAMPLES -g ${REF}.fai -empty -i $BEDGZ | bgzip > ALL.bg.gz
tabix -f -p bed -S 1 ALL.bg.gz
Command line magic
The code above works as follows. We first use csvtk to grep (search) for the population names red and yellow in the Taxon column in sampleinfo.csv, thereby filtering the output to lines where Taxon matches the population names. Then, we cut out the interesting column SampleAlias and remove the header (grep -v SampleAlias matches anything but SampleAlias). Finally, tr translates newline character \n to space. The output is stored in the SAMPLES variable through the command substitution ($()) syntax.
We then iterate through the $SAMPLES to generate the input file names with the echo command, storing the output in $BEDGZ. These variables are passed on to bedtools unionbedg to generate a bedgraph file combining all samples.
As mentioned previously, we also need to combine coverages per populations yellow and red.
Total coverage
Since we eventually want to filter on total coverage, we sum per sample coverages for each sample set with awk:
bgzip -c -d ALL.bg.gz | \
awk -v FS="\t" -v OFS="\t" 'NR > 1 {sum=0; for (i=4; i<=NF; i++) sum+=$i; print $1, $2, $3, sum}' | \
bgzip > ALL.sum.bed.gz
tabix -f -p bed ALL.sum.bed.gzHere we use awk to sum from columns 4 and up (NF is the number of the last column).
For illustration, we plot the total coverage:
bedtools intersect -a <(bedtools makewindows -g ${REF}.fai -w 1000) \
-b ALL.sum.bed.gz -wa -wb | \
bedtools groupby -i - -g 1,2,3 -c 7 -o mean | \
csvtk plot -t line -x 2 -y 4 --point-size 0.01 --xlab Position \
--ylab Coverage --width 9.0 --height 3.5 > fig-plot-total-coverage.png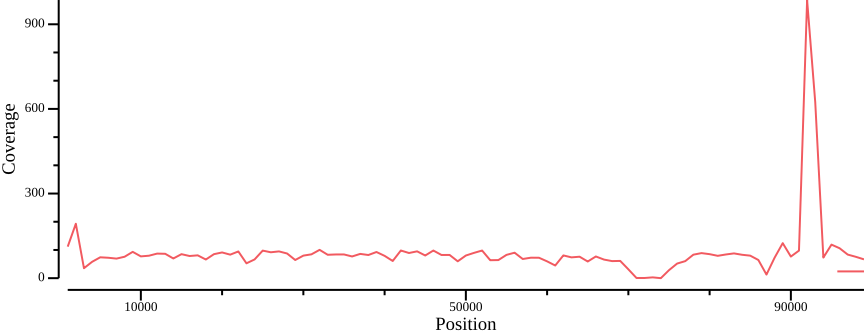
In order to define thresholds for subsequent filtering, we need to know the total coverage distribution. Therefore, we plot the proportion of the genome coverage versus depth of coverage (similar to k-mer plots in sequence assembly projects). To do so we must summarize our coverage file such that we count how many bases have a given coverage. This can be achieved by noting that each row in the BED file consists of the columns CHROM, START, END, and COVERAGE. We can generate a histogram table by, for each value of COVERAGE, summing the length of the regions (END - START).
# Add column containing length of region (end - start)
csvtk mutate2 -t -H -w 0 -e '$3-$2' ALL.sum.bed.gz | \
# Sum the regions and *group* by the coverage (fourth column);
# this gives the total number of bases with a given coverage
csvtk summary -t -H -g 4 -f 5:sum -w 0 | \
csvtk sort -t -k 1:n | \
awk -v cumsum=0 'BEGIN {OFS=","; cumsum=0} {cumsum += $2; print $1,$2,cumsum}' > ALL.sum.bed.csvWe plot the coverage distribution below, along with a plot of the cumulative coverage.
csvtk plot line -H ALL.sum.bed.csv -x 1 -y 2 --point-size 0.01 \
--xlab "Depth of coverage (X)" --ylab "Genome coverage (bp)" \
--width 9.0 --height 3.5 > fig-plot-total-coverage-distribution.png
csvtk plot line -H ALL.sum.bed.csv -x 1 -y 3 --point-size 0.01 \
--xlab "Depth of coverage (X)" --ylab "Cumulative genome coverage (kbp)" \
--width 9.0 --height 3.5 > fig-plot-total-coverage-distribution-cumulative.png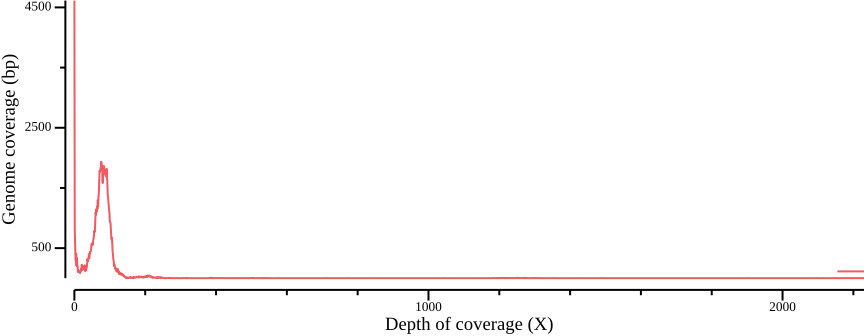
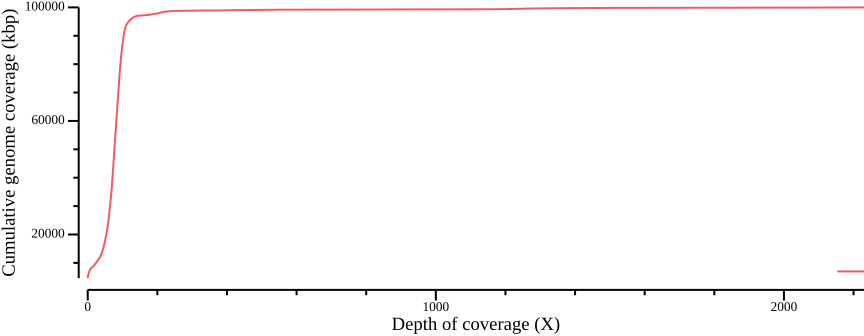
In Figure 12 a, a diploid peak is evident at around coverage X=100; we zoom in on that region to get a better view:
csvtk plot line -H ALL.sum.bed.csv -x 1 -y 2 --point-size 0.01 \
--xlab "Depth of coverage (X)" --ylab "Genome coverage (bp)" \
--width 9.0 --height 3.5 --x-min 40 --x-max 140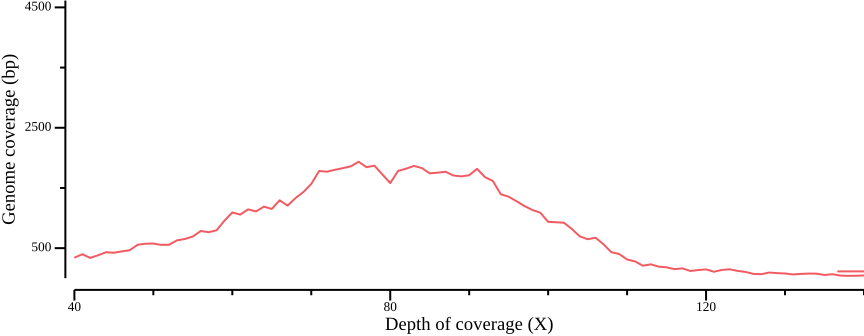
(Lou et al., 2021) point out that appropriate thresholds depend on the data set, but as a general rule recommend a minimum depth threshold at <0.8X average coverage, and a maximum depth threshold at mean coverage plus one or two standard deviations. For the sake of simplicity, you could here infer a cutoff simply by manually inspecting Figure 13; here, we will use the range 50-110.
We then use these thresholds to generate a BED file containing regions that are accessible, i.e., have sufficient coverage for downstream analyses. We also calculate the number of bases that pass the filtering criteria.
csvtk filter -t -H ALL.sum.bed.gz -f '4>50' | \
csvtk filter -t -H -f '4<110' | \
bgzip -c > ALL.sum.depth.bed.gz
bedtools genomecov -i ALL.sum.depth.bed.gz -g ${REF}.fai | grep genomeConsequently, 75.2% of the genome is accessible by depth.
Exercise
Generate coverage sums for the red and yellow sample sets, and from these determine coverage thresholds and apply the thresholds to generate BED files with accessible regions.
Answer
We base the answer on the previous code.
for pop in red yellow; do
bgzip -c -d $pop.bg.gz | \
awk -v FS="\t" -v OFS="\t" 'NR > 1 {sum=0; for (i=4; i<=NF; i++) sum+=$i; print $1, $2, $3, sum}' | \
bgzip > $pop.sum.bed.gz
tabix -f -p bed $pop.sum.bed.gz
csvtk mutate2 -t -H -w 0 -e '$3-$2' $pop.sum.bed.gz | \
csvtk summary -t -H -g 4 -f 5:sum -w 0 | \
csvtk sort -t -k 1:n | \
awk -v cumsum=0 'BEGIN {OFS=","; cumsum=0} {cumsum += $2; print $1,$2,cumsum}' > $pop.sum.bed.csv
donefor pop in red yellow; do
cat ${pop}.sum.bed.csv | \
csvtk plot line -x 1 -y 2 --point-size 0.01 \
--xlab "Depth of coverage (X)" --ylab "Genome coverage (bp)" \
--width 9.0 --height 3.5 --x-min 0 --x-max 100 > \
fig-plot-total-coverage-distribution-hist-zoom-in-$pop.png
done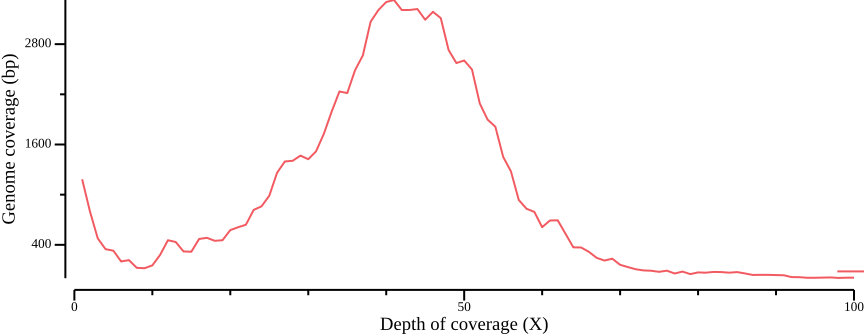
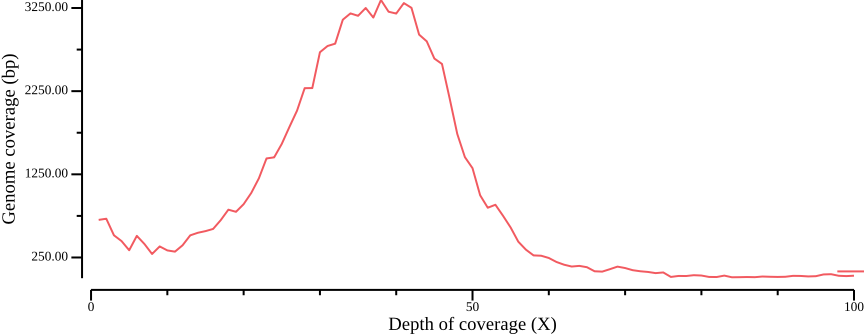
Based on Figure 14, we generate bed files with depths passing cutoffs:
# red filter: 20-60
csvtk filter -t -H red.sum.bed.gz -f '4>20' | \
csvtk filter -t -H -f '4<60' | \
bgzip -c > red.sum.depth.bed.gz
bedtools genomecov -i red.sum.depth.bed.gz -g ${REF}.fai | grep genome
# yellow filter: 20-55
csvtk filter -t -H yellow.sum.bed.gz -f '4>20' | \
csvtk filter -t -H -f '4<55' | \
bgzip -c > yellow.sum.depth.bed.gz
bedtools genomecov -i yellow.sum.depth.bed.gz -g ${REF}.fai | grep genomegenome 0 20853 100000 0.20853
genome 1 79147 100000 0.79147
genome 0 23231 100000 0.23231
genome 1 76769 100000 0.76769Now we have combined total per sample coverage for ALL samples, and for sample sets red and yellow. The upcoming task will be to generate sequence masks from the total coverage and minimum number of individuals with coverage greater than zero.
Filter on minimum number of individuals
In addition to filtering on total coverage, we will also filter on the minimum number of individuals with a minimum depth. This is to account for cases where regions that pass the minimum coverage filter originate from just a few samples with unusually high coverage. Here, we will remove sites where more than 50% of individuals have zero coverage.
bgzip -c -d ALL.bg.gz | \
awk -v FS="\t" 'BEGIN {OFS="\t"} NR > 1 {count=0; for (i=4; i<=NF; i++) {if ($i>0) count+=1}; if (count>=((NF-3)*0.5)) {print $1, $2, $3}}' | \
bgzip > ALL.ind.bed.gz
tabix -f -p bed ALL.ind.bed.gzSequence masks
Finally, as before, we can convert the BED output to sequence mask files in FASTA format. Recall that we first have to make a template genome mask file where all positions are masked:
awk 'BEGIN {OFS="\t"} {print $1, 0, $2}' ${REF}.fai > ${REF}.bed
bedtools maskfasta -fi ${REF} -mc 1 -fo ${REF}.mask.fa -bed ${REF}.bedThen, for each sample set, we will use bedtools intersect to intersect the BED files corresponding to the total sum coverage and the filter on number of individuals. bedtools intersect makes it easy to combine multiple BED files, so any other filters, or genomic features such as exons, could be added to make a compound mask file. The resulting BED files is used as input to bedtools maskfasta.
bedtools intersect -a ALL.sum.depth.bed.gz -b ALL.ind.bed.gz \
-g ${REF}.fai | bgzip > ALL.unmask.bed.gz
tabix -f -p bed ALL.unmask.bed.gz
bedtools maskfasta -fi ${REF}.mask.fa -mc 0 -fo ${REF}.unmask.fa \
-bed ALL.unmask.bed.gz
head -n 3 ${REF}.unmask.fa>LG4
111111111111111111111111111111111111111111111111111111111111
111111111111111111100000000000000000000000000000000111111111We can once again convince ourselves that this has worked by counting the number of unmasked positions:
# tr: -d deletes all characters not (-c, complement) in the character
# set '0'. wc: -m option counts characters
cat ${REF}.unmask.fa | tr -d -c '0' | wc -m
bedtools genomecov -i ALL.unmask.bed.gz -g ${REF}.fai | grep genome75236
genome 0 24764 100000 0.24764
genome 1 75236 100000 0.75236Note that 0 and 1 in the bedtools genomecov output refers to coverage (i.e., absence/presence) and not unmask/mask as in the mask FASTA file.
Conclusion
Congratulations! You have now gone through a set of tedious and complex steps to generate output files that determine what regions in a reference DNA sequence are amenable to analysis. In the next exercise we will use these files as inputs to different programs that calculate diversity statistics from population genomic data.
References
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., Handsaker, R. E., Lunter, G., Marth, G. T., Sherry, S. T., McVean, G., & Durbin, R. (2011). The variant call format and VCFtools. Bioinformatics (Oxford, England), 27(15), 2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., Whitwham, A., Keane, T., McCarthy, S. A., Davies, R. M., & Li, H. (2021). Twelve years of SAMtools and BCFtools. GigaScience, 10(2), giab008. https://doi.org/10.1093/gigascience/giab008
Garrison, E., Kronenberg, Z. N., Dawson, E. T., Pedersen, B. S., & Prins, P. (2022). A spectrum of free software tools for processing the VCF variant call format: Vcflib, bio-vcf, Cyvcf2, hts-nim and slivar. PLOS Computational Biology, 18(5), e1009123. https://doi.org/10.1371/journal.pcbi.1009123
Hou, H., Pedersen, B., & Quinlan, A. (2021). Balancing efficient analysis and storage of quantitative genomics data with the D4 format and D4tools. Nature Computational Science, 1(6), 441–447. https://doi.org/10.1038/s43588-021-00085-0
Li, H. (2014). Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics, 30(20), 2843–2851. https://doi.org/10.1093/bioinformatics/btu356
Lou, R. N., Jacobs, A., Wilder, A. P., & Therkildsen, N. O. (2021). A beginner’s guide to low-coverage whole genome sequencing for population genomics. Molecular Ecology, 30(23), 5966–5993. https://doi.org/10.1111/mec.16077
Maruki, T., & Lynch, M. (2017). Genotype Calling from Population-Genomic Sequencing Data. G3 Genes|Genomes|Genetics, 7(5), 1393–1404. https://doi.org/10.1534/g3.117.039008
Nielsen, R., Paul, J. S., Albrechtsen, A., & Song, Y. S. (2011). Genotype and SNP calling from next-generation sequencing data. Nature Reviews Genetics, 12(6), 443–451. https://doi.org/10.1038/nrg2986
Pedersen, B. S., & Quinlan, A. R. (2018). Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics, 34(5), 867–868. https://doi.org/10.1093/bioinformatics/btx699
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., Maller, J., Sklar, P., de Bakker, P. I. W., Daly, M. J., & Sham, P. C. (2007). PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. The American Journal of Human Genetics, 81(3), 559–575. https://doi.org/10.1086/519795
Quinlan, A. R., & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26(6), 841–842. https://doi.org/10.1093/bioinformatics/btq033
Shen, W., Le, S., Li, Y., & Hu, F. (2016). SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLOS ONE, 11(10), e0163962. https://doi.org/10.1371/journal.pone.0163962
Footnotes
The password is provided by the course instructor↩︎
This exercise is inspired by and based on https://speciationgenomics.github.io/filtering_vcfs/]↩︎
We select a fix number that should contain enough information to generate reliable statistics. This number should not change significantly even when files contain vastly different numbers of sites, which is why we need adjust the parameter r to the number of sites in the file.↩︎
vcftoolsdoes not have a-hor--helpoption.↩︎The
2>/dev/nulloutputs messages fromvcftoolsto a special file/dev/nullwhich is a kind of electronic dustbin.↩︎On viewing
csvtkplots: either you can redirect (>) the results fromcsvtkto a png output file, or you can pipe (|) it to the commanddisplay(replace> $OUT.ldepth.pngby| display, which should fire up a window with your plot.↩︎You can try different values of the
--binsoption↩︎Purcell et al. (2007), p. 565 gives a coherent derivation of this estimator↩︎
We need to generate a BED file representation of the FASTA index unfortuanely as
bedtools makefastadoesn’t handle FASTA indices natively.↩︎For
vcftools; unfortunate, but that’s the way it is↩︎The one-liner combines the results of several commands in a pipe stream. Also, Bash redirections are used to gather the results from the output of
bedtools makewindowstobedtools intersect. The intersection commands collects coverage data in 1kb windows that are then summarized bybedtools groupby.↩︎The
%operator deletes the shortest match of$substringfrom back of$string:${string%substring}. See Bash string manipulation for more information.↩︎Instead of using the command substitution, you could look into the sample info file and set the sample names manually:
SAMPLES="PUN-Y-BCRD PUN-R-ELF PUN-Y-INJ PUN-R-JMC PUN-R-LH PUN-Y-LO PUN-R-MT PUN-Y-PCT PUN-Y-POTR PUN-R-UCSD"↩︎