
Variant calling
From sequence data to variant call set
Per Unneberg
NBIS
15-Nov-2023
Yesterday
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T | T | A | C | A | A | T | C | C | G | A | T | C | G | T |
| T | T | A | C | G | A | T | G | C | G | C | T | C | G | T |
| T | C | A | C | A | A | T | G | C | G | A | T | G | G | A |
| T | T | A | C | G | A | T | G | C | G | C | T | C | G | T |
| * | * | * | * | * | * |
\[ \begin{align} \pi & = \sum_{j=1}^S h_j = \sum_{j=1}^{S} \frac{n}{n-1}\left(1 - \sum_i p_i^2 \right) \\ & \stackrel{S=6,\\ n=4}{=} \sum_{j=1}^{6} \frac{4}{3}\left(1 - \sum_i p_i^2\right) \\ & = \frac{4}{3}\left(\mathbf{\color{#a7c947}{4}}\left(1-\frac{1}{16}- \frac{9}{16}\right) + \mathbf{\color{#a7c947}{2}}\left(1 - \frac{1}{4} - \frac{1}{4}\right)\right) = \frac{10}{3} \end{align} \]

\[ \begin{align} \pi & = \frac{\sum_{i=1}^{n-1}i(n-i)\xi_i}{n(n-1)/2} \\ & \stackrel{n=4}{=} \frac{1*(4-1)*4 + 2*(4-2)*2}{6} = \frac{10}{3} \end{align} \]
This is not how real data looks like from the beginning…
The process

Variant and genotype calling
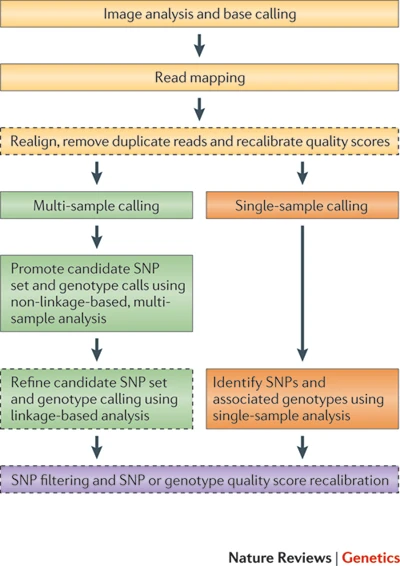
Nielsen et al. (2011)
- SNP calling
- Identification of polymorphic sites (\(>1\)% allele frequency)
- Variant calling
- Identification of variant sites (sufficient that any allele differs)
- Genotype calling
- Determine the allele combination for each individual (aa, aA, or AA for bi-allelic variants)
Knowing variant sites informs us of possible genotypes and improves genotyping.
Example: knowing a site has A or C limits possible genotype calls to AA, AC, or CC
Sequencing technologies

![]()
Illumina NovaSeq 600
Scale up and down with a tunable output of up to 6 Tb and 20B single reads in < 2 days.
Up to 2X250 bp read length. Price example: 8,000 SEK total for resequencing 3Gbp genome to 30X
PacBio Revio
Up to 360 Gb of HiFi reads per day, equivalent to 1,300 human whole genomes per year.
Tens of kilobases long HiFi reads. Price example (Sequel II): ~35kSEK per library and SMRT cell
DNA sequence quality control
Quality values represent the probability \(P\) that the call is incorrect. They are coded as Phred quality scores \(Q\). Here, \(Q=20\) implies 1% probability of error, \(Q=30\) 0.1% and so on. Typically you should not rely on quality values below \(20\).
\[ Q = -10 \log_{10} P \]

Sequencing approaches

Despite price drop, still need to make choices regarding depth and breadth of sequencing coverage and number of samples.

Genome assembly and population resequencing

Population resequencing

Sequence alignment maps reads to a reference
. characters is the consensus sequence. Bases are colored by nucleotide. Letter case indicates forward (upper-case) or reverse (lower-case) alignment. * is placeholder for deleted base.
Aim of sequence alignment (read mapping) is to determine source in reference sequence. Some commonly used read mappers for resequencing are
- BWA, BWA-MEM (H. Li, 2013)
- Novoalign (https://www.novocraft.com/)
- Minimap2 (H. Li, 2018)
For a recent comprehensive comparison see Donato et al. (2021)
GATK best practice

Pros
- Best practices
- Large documentation
- Variant quality score recalibration
Cons
- Human-centric - very slow runtime on genomes with many sequences
- Complicated setup
The monkeyflower system

From https://jgi.doe.gov/csp-2021-genomic-resources-for-mimulus/
Plants in the genus Mimulus inhabit highly variable habitats and are famous for their extraordinary ecological diversity. Mimulus is now a powerful system for ecological genomic studies, thanks to its experimental tractability, rapidly growing research community, and the JGI-generated reference genome for M. guttatus.