SN 0 number of samples: 10
SN 0 number of records: 12673
SN 0 number of no-ALTs: 0
SN 0 number of SNPs: 10403
SN 0 number of MNPs: 0
SN 0 number of indels: 2291
SN 0 number of others: 0
SN 0 number of multiallelic sites: 1042
SN 0 number of multiallelic SNP sites: 210Variant filtering
From raw variant calls to high-quality call sets
Per Unneberg
NBIS
15-Nov-2023
Why we need to filter variants

Error rate of variant calls (SNPs and INDELs) largely unknown. Two major sources of error are
- erroneous realignment in low-complexity regions
- incomplete reference sequence

Li (2014)
What about machine learning?
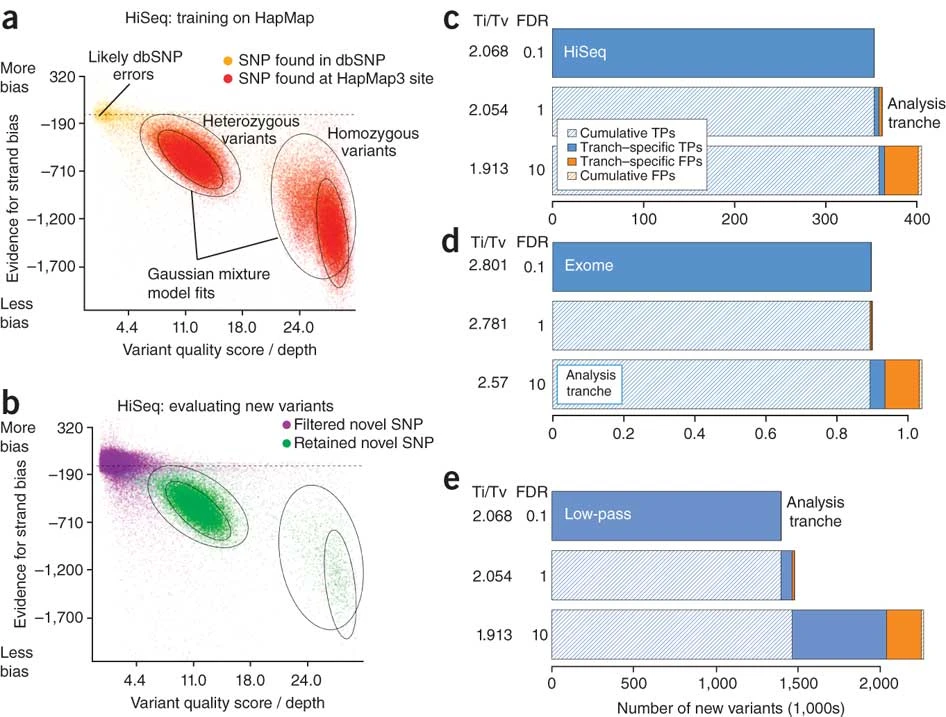
DePristo et al. (2011)
Variant Quality Score Recalibration
Motivation: look at context statistics and integrate over multiple dimensions
- training data: subset of known variants (from validated resources, e.g. 1000 Genomes)
- compile multiple statistics (allele depth, read count, quality, …)
- fit Gaussian mixture model
- reassign quality scores to variant call set
Caveat: database of known variants often not known for non-model organisms.
Mean depth and variant quality distribution
Code
vcf <- "variantsites.vcf.gz"
system(paste("vcftools --gzvcf", vcf, "--site-depth 2>/dev/null"))
data <- read.table("out.ldepth", header = TRUE)
x <- as.data.frame(table(data$SUM_DEPTH))
lower <- 0.8 * median(data$SUM_DEPTH)
upper <- median(data$SUM_DEPTH) + 1 * sd(data$SUM_DEPTH)
xupper <- ceiling(upper/100) * 100
ggplot(x, aes(x = as.numeric(as.character(Var1)), y = Freq)) + geom_line() + xlab("Depth") +
ylab("bp") + xlim(0, xupper) + geom_vline(xintercept = lower, color = "red",
size = 1.3) + geom_vline(xintercept = upper, color = "red", size = 1.3) + ggtitle("Example threshold: 0.8X median depth, median depth + 2sd")
Depth uneven. High coverage often repetitive sequence. Too low coverage will bias SNP calling due to undersampling of alleles.
Code
system(paste("vcftools --gzvcf", vcf, "--site-quality 2>/dev/null"))
data <- read.table("out.lqual", header = TRUE)
ggplot(subset(data, QUAL < 1000), aes(x = QUAL)) + geom_histogram(fill = "white",
color = "black", bins = 50) + xlab("Quality value") + ylab("Count") + geom_vline(xintercept = 30,
color = "red", size = 1.3) + ggtitle("Example threshold: Q30")
Filter variants with too low quality (Q30=0.001% chance of being wrong)
Missing data per individual and site
Code

Missing number of sites per individual. Too many would indicate poor sample quality.
Code
system(paste("vcftools --gzvcf", vcf, "--missing-site 2>/dev/null"))
data <- read.table("out.lmiss", header = TRUE)
ggplot(data, aes(x = F_MISS)) + geom_histogram(fill = "white", color = "black", bins = 10) +
xlab("F_MISS") + ylab("Count") + geom_vline(xintercept = 0.25, color = "red",
size = 1.3) + ggtitle("Missing data per site: example threshold F_MISS=0.25")
Fraction missing calls per site. Could warrant separate filters when comparing populations (e.g., total missing 0.2, but population A has 0.1 missing, population B 0.4).
Minor allele frequency and heterozygosity
Code
system(paste("vcftools --gzvcf", vcf, "--freq2 --max-alleles 2 2>/dev/null"))
data <- read.table("out.frq", skip = 1)
colnames(data) <- c("CHROM", "POS", "N_ALLELES", "N_CHR", "FREQ1", "FREQ2")
data$MAF <- apply(data, 1, function(x) as.numeric(min(x[5], x[6])))
ggplot(data, aes(x = MAF)) + geom_histogram(fill = "white", color = "black", bins = 10) +
xlab("MAF") + ylab("Count") + geom_vline(xintercept = 0.1, color = "red", size = 1.3) +
ggtitle("Minor allele frequency: example threshold MAF=0.1")
n=12; mutations 0, 4, 5 (red) are singletons and would fail MAF<=0.1
Reasonable cutoff 0.05-0.1 for PCA, population structure.
But! Statistics based on diversity or the SFS should not be filtered on MAF
Code

- F=0: Hardy-Weinberg Equilibrium
- F>0: deficit of heterozygotes; inbreeding, Wahlund effect (population substructure), allele dropout
- F<0: surplus of heterozygotes; could be sample contamination, poor sequence quality (mismapping)
Coverage tracks and sequence masks

Coverage tracks and sequence masks

Coverage tracks and sequence masks

Coverage tracks and sequence masks

Coverage tracks and sequence masks

Coverage tracks and sequence masks

Coverage tracks and sequence masks

Coverage tracks and sequence masks
