
Simulation
Primer on the coalescent and forward simulation
Per Unneberg
NBIS
15-Nov-2023
The Wright-Fisher model and simulations


Mean reproductive success = 63.4%. Can show for large populations P(no descendants)=\(1 - e^{-1} \approx 0.632\)
Forward and backward simulation


Simulated nodes are filled with black. Genealogy of interest is highlighted in thick black lines.
Coalescent simulations
The coalescent simulates the genealogy of a sample of individuals on which mutations are “sprinkled” according to a Poisson process.
- Simulate ancestry (genealogy)

Coalescent simulations
The coalescent simulates the genealogy of a sample of individuals on which mutations are “sprinkled” according to a Poisson process.
- Simulate ancestry (genealogy)
- Simulate mutations

Coalescent simulations
The coalescent simulates the genealogy of a sample of individuals on which mutations are “sprinkled” according to a Poisson process.
- Simulate ancestry (genealogy)
- Simulate mutations
Exercise
How many mutations are common to all samples? How many mutations does sample 1 have? Sample 2?
Assuming the ancestral state is denoted 0 (prior to the first generation) and the derived state 1, what are the sequences of the samples?

Simulating genealogies (Hahn, 2019, p. 115)
- Start with \(i=n\) chromosomes

Simulating genealogies (Hahn, 2019, p. 115)
Start with \(i=n\) chromosomes
Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)

Simulating genealogies (Hahn, 2019, p. 115)
Start with \(i=n\) chromosomes
Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)
Choose two chromosomes at random to coalesce

Simulating genealogies (Hahn, 2019, p. 115)
Start with \(i=n\) chromosomes
Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)
Choose two chromosomes at random to coalesce
Merge the two lineages and set \(i \rightarrow i - 1\)

Simulating genealogies (Hahn, 2019, p. 115)
- Start with \(i=n\) chromosomes
- Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)
- Choose two chromosomes at random to coalesce
- Merge the two lineages and set \(i \rightarrow i - 1\)
- If \(i>1\), go to step 2; if not, stop.

Simulating genealogies (Hahn, 2019, p. 115)
- Start with \(i=n\) chromosomes
- Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)
- Choose two chromosomes at random to coalesce
- Merge the two lineages and set \(i \rightarrow i - 1\)
- If \(i>1\), go to step 2; if not, stop.

Simulating genealogies (Hahn, 2019, p. 115)
- Start with \(i=n\) chromosomes
- Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)
- Choose two chromosomes at random to coalesce
- Merge the two lineages and set \(i \rightarrow i - 1\)
- If \(i>1\), go to step 2; if not, stop.

Simulating genealogies (Hahn, 2019, p. 115)
- Start with \(i=n\) chromosomes
- Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)
- Choose two chromosomes at random to coalesce
- Merge the two lineages and set \(i \rightarrow i - 1\)
- If \(i>1\), go to step 2; if not, stop.

Simulating genealogies (Hahn, 2019, p. 115)
- Start with \(i=n\) chromosomes
- Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)
- Choose two chromosomes at random to coalesce
- Merge the two lineages and set \(i \rightarrow i - 1\)
- If \(i>1\), go to step 2; if not, stop.

Simulating genealogies (Hahn, 2019, p. 115)
- Start with \(i=n\) chromosomes
- Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)
- Choose two chromosomes at random to coalesce
- Merge the two lineages and set \(i \rightarrow i - 1\)
- If \(i>1\), go to step 2; if not, stop.

Simulating genealogies (Hahn, 2019, p. 115)
- Start with \(i=n\) chromosomes
- Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)\(^1\)
- Choose two chromosomes at random to coalesce
- Merge the two lineages and set \(i \rightarrow i - 1\)
- If \(i>1\), go to step 2; if not, stop.
\(^1\): The exponential can be parametrized in two different ways, so that the parameter to the function is either \(\lambda\) or \(\beta=1/\lambda\).

Simulating genealogies (Hahn, 2019, p. 115)
- Start with \(i=n\) chromosomes
- Choose time to next coalescent event from an exponential distribution with parameter \(\lambda=i(i-1)/2\)\(^1\)
- Choose two chromosomes at random to coalesce
- Merge the two lineages and set \(i \rightarrow i - 1\)
- If \(i>1\), go to step 2; if not, stop.
\(^1\): The exponential can be parametrized in two different ways, so that the parameter to the function is either \(\lambda\) or \(\beta=1/\lambda\).

Some properties of the tree

Expected waiting time to coalesce when \(i\) lineages: \(E(T_i) = \frac{2}{i(i-1)}\)
Branch lengths can be derived from waiting times. For instance, \(\tau_1=\tau_2=T_5+T_4\) and \(\tau_4=\tau_5=T_5\)
Time to the most recent common ancestor (MRCA) is sum of wating times: \(T_{MRCA} = \sum_{i=2}^n T_i\)
with expected value \(E(T_{MRCA}) = \sum_{i=2}^nE(T_i) = 2\left(1 - \frac{1}{n}\right)\)
The expected total tree height is \(E(T_{total}) = \sum_{i=2}^n iE(T_i) = 2\sum_{i=2}^n\frac{1}{i-1}\)
Diminishing returns of adding more samples

Adding mutations

Mutations are added by placing them on branches, where the probability of ending up on a branch \(\tau_i\) is equal to its normalized length, where normalization is by the total tree branch length:
\[ P(\text{mutation on branch }i) = \frac{\tau_i}{\sum_j\tau_j} = \frac{\tau_i}{T_{total}} \]
The total number of segregating sites \(S\) to be thrown on the tree is modelled as a Poisson random variable which expresses the probability of a given number of events in time \(t\):
\[ S = Po(\theta/2T_{total}) \]
The coalescent and diversity
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T | T | A | C | A | A | T | C | C | G | A | T | C | G | T |
| T | T | A | C | G | A | T | G | C | G | C | T | C | G | T |
| T | C | A | C | A | A | T | G | C | G | A | T | G | G | A |
| T | T | A | C | G | A | T | G | C | G | C | T | C | G | T |
| * | * | * | * | * | * |
\[ \begin{align} \pi & = \sum_{j=1}^S h_j = \sum_{j=1}^{S} \frac{n}{n-1}\left(1 - \sum_i p_i^2 \right) \\ & \stackrel{S=6,\\ n=4}{=} \sum_{j=1}^{6} \frac{4}{3}\left(1 - \sum_i p_i^2\right) \\ & = \frac{4}{3}\left(\mathbf{\color{#a7c947}{4}}\left(1-\frac{1}{16}-\frac{9}{16}\right) + \mathbf{\color{#a7c947}{2}}\left(1 - \frac{1}{4} - \frac{1}{4}\right)\right) = \frac{10}{3} \end{align} \]


In this notation one can show that \(\pi\), the nucleotide diversity, is
\[ \begin{align} \pi & = \frac{\sum_{i=1}^{n-1}i(n-i)\xi_i}{n(n-1)/2} \\ & \stackrel{n=4}{=} \frac{1*(4-1)*4 + 2*(4-2)*2}{6} = \frac{10}{3} \end{align} \]
Many statistical quantities can be related to the site frequency spectrum (SFS), which is a summary of the frequencies of the segregating sites. Let \(\xi_i\) be the number of chromosomes in the sample with \(i\) minor alleles. In the example above we have \(S=6\) mutations on \(n=4\) chromosomes.
The impact of topology on the SFS

On non-recombining chromosomes and assortment

Both siblings inherit chromosome from paternal grandfather
Chromosomes coalesce at father

Siblings inherit different grandparental chromosomes \(\Rightarrow\) chromosomes coalesce God knows when in the past
Genealogies differ
The ancestral recombination graph



Y. C. Brandt et al. (2022, fig. 1a)
Properties:
- marginal trees constitute a sequence of trees (tree sequence) along a chromosome
- each tree represents the genealogy of a non-recombining part of the chromosome
- neighbouring trees are correlated
Interpretation: chromosomes are mosaics of non-recombining units
The ancestral recombination graph as sequence of trees

Properties:
- marginal trees constitute a sequence of trees (tree sequence) along a chromosome
- each tree represents the genealogy of a non-recombining part of the chromosome
- neighbouring trees are correlated
Interpretation: chromosomes are mosaics of non-recombining units
msprime stores variation data as tree sequences

Tree sequences (Baumdicker et al., 2022, fig. 2)
Tree sequences compress data

Data compression (Kelleher et al., 2019, fig. 1c)
SLiM
SLiM (Selection on Linked Mutations) (Haller & Messer, 2019) is a forwards-time simulator. As its name implies, it models selection and thus is a complement to coalescent-based simulators.
Why SLiM?
- flexibility - scripting language Eidos allows for modelling complex scenarios with little code
- performance - optimized code base
- GUI - interactive execution and graphical debugging
Haller & Messer (2022)

Forward simulation in SLiM
- Execution of
first()events - Execution of
early()events - Generation of offspring; for each offspring:
- Choose source subpop for parental individuals, based on migration rates
- Choose parent 1, based on cached fitness values
- Choose parent 2, based on fitness and any defined
mateChoice()callbacks - Generate the candidate offspring, with mutation and recombination (including
mutation()andrecombination()callbacks) - Suppress/modify the candidate, using defined
modifyChild()callbacks
- Removal of fixed mutations unless
convertToSubstitution==F - Offspring become parents
- Execution of
late()events - Fitness recalculation using
mutationEffect()andfitnessEffect()callbacks - Tick/cycle count increment

Recapitation - combining the best of two worlds
A recent addition to SLiM is that it records data in tree sequence format (as in msprime) we can combine backward and forward simulations!
Backward simulation that adds coalescent (neutral) history

Forward simulation with selection or some other process that isn’t supported by the coalescent

Simulation of null distributions

Authors inferred demographic history and recombination map. The inferred values were used to simulate data with msprime. SweepFinder2 (DeGiorgio et al., 2016) was used to calculate composite likelihood ratio (CLR) scores on both observed and simulated data to assess significance quantiles.
Simulations of genomic landscapes in monkeyflower
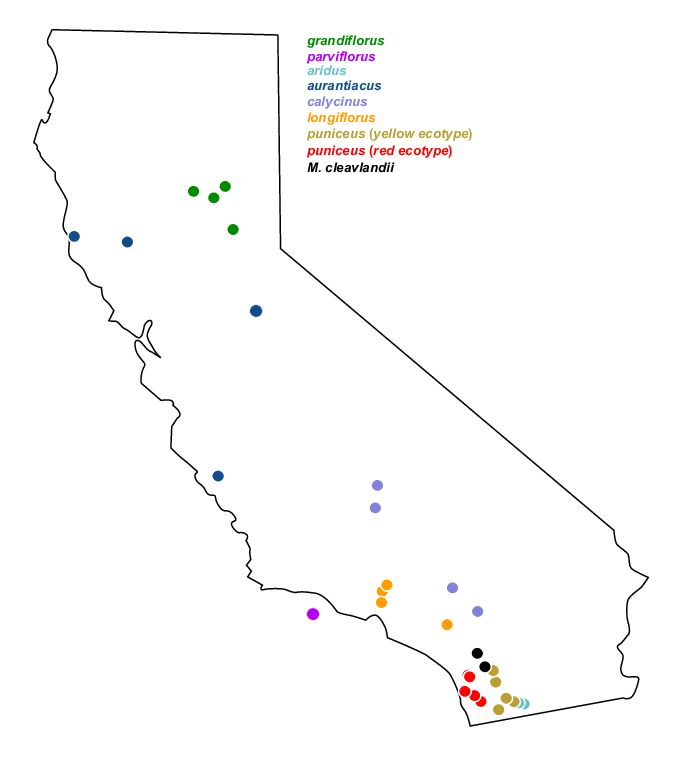
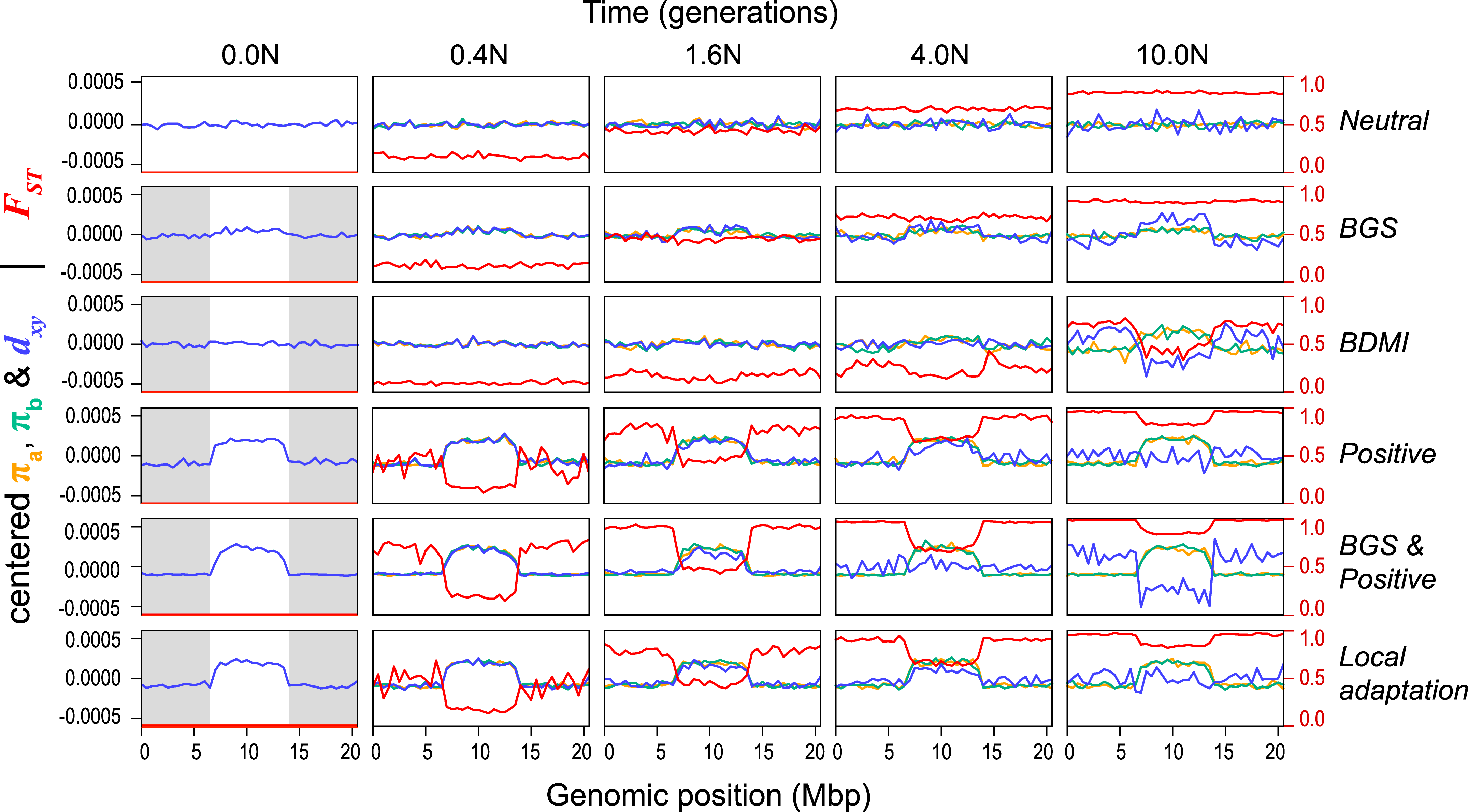
Monkeyflower (Mimulus aurantiacus) radiation in California shows wide adaptive range and phenotypic diversity. Stankowski et al. (2019) et al use simulations (SLiM and msprime) to shed light on processes that shape genomic landscapes, which are correlated between species.
We will look more closely at this system tomorrow.
Neural network recombination landscape prediction

Use msprime simulations to generate training data for neural network.