mkdir -p monkeyflower
cd monkeyflowerGenetic diversity landscapes
Investigating the diversity landscape in Monkeyflower
Compute environment setup
If you haven’t already done so, please read Compute environment for information on how to prepare your working directory.
In this exercise we will look at measures that describe variation and compile statistics along a sequence. By scanning variation in windows along the sequence (a.k.a. genomic scan) we can identify outlier regions whose pattern of variation could potentially be attributed to causes other than neutral processes, such as adaptation or migration. We will use the Monkeyflower system to generate a diversity landscape.
Important
The commands of this document have been run on a subset (a subregion) of the data. Therefore, although you will use the same commands, your results will differ from those presented here.
Learning objectives
- describe and calculate commonly used measures of variation, including nucleotide diversity \(\pi\), divergence \(d_{XY}\) and differentiation \(F_{ST}\)
- perform genome scans of diversity and plot the results
- identify outlier regions of interest
- untangle processes that drive patterns of variation using simulated data
Data setup
Move to your course working directory /proj/naiss2023-22-1084/users/USERNAME, create an exercise directory called monkeyflower and cd to it:
cd /proj/naiss2023-22-1084/users/USERNAME
mkdir -p monkeyflower
cd monkeyflowerRetrieve the data with rsync. You can use the --dry-run option to see what is going to happen; just remove it when you are content:
rsync --dry-run -av \
/proj/naiss2023-22-1084/webexport/monkeyflower/LG4/* .
# Remove the dry run option to retrieve data. The dot is important!
rsync --dry-run -av /proj/naiss2023-22-1084/webexport/monkeyflower/LG4/* .Create an exercise directory called monkeyflower and cd to it:
Retrieve the variant files from https://export.uppmax.uu.se/naiss2023-22-1084/monkeyflower/LG4 with wget1:
wget --user pgip --password PASSWORD \
--recursive --accept='*.*' \
--reject='*.gif','index*' \
--no-parent --no-directories \
--no-clobber \
--directory-prefix=. \
https://export.uppmax.uu.se/naiss2023-22-1084/monkeyflower/LG4/
Tools
Execute the following command to load modules:
module load uppmax bioinfo-tools \
bcftools/1.17 \
BEDTools/2.29.2 \
vcftools/0.1.16 \
pixy/1.2.5.beta1csvtk has been added to the module system and can be loaded as follows:
module use /proj/naiss2023-22-1084/modules
module load csvtkCopy the contents to a file environment.yml and install packages with mamba env update -f environment.yml.
channels:
- conda-forge
- bioconda
- default
dependencies:
- bcftools=1.15.1
- bedtools=2.31.0
- csvtk=0.28.0
- vcftools=0.1.16There is no pixy Conda package for Python >= 3.9, so it must be manually installed with pip:
python -m pip install git+https://github.com/ksamuk/pixy.gitPreparation
In this exercise, we will be analysing the full data set for linkage group 4 (LG4), consisting of all 37 samples.
| Filename | records | samples |
|---|---|---|
| allsites.vcf.gz | 1e+05 | 37 |
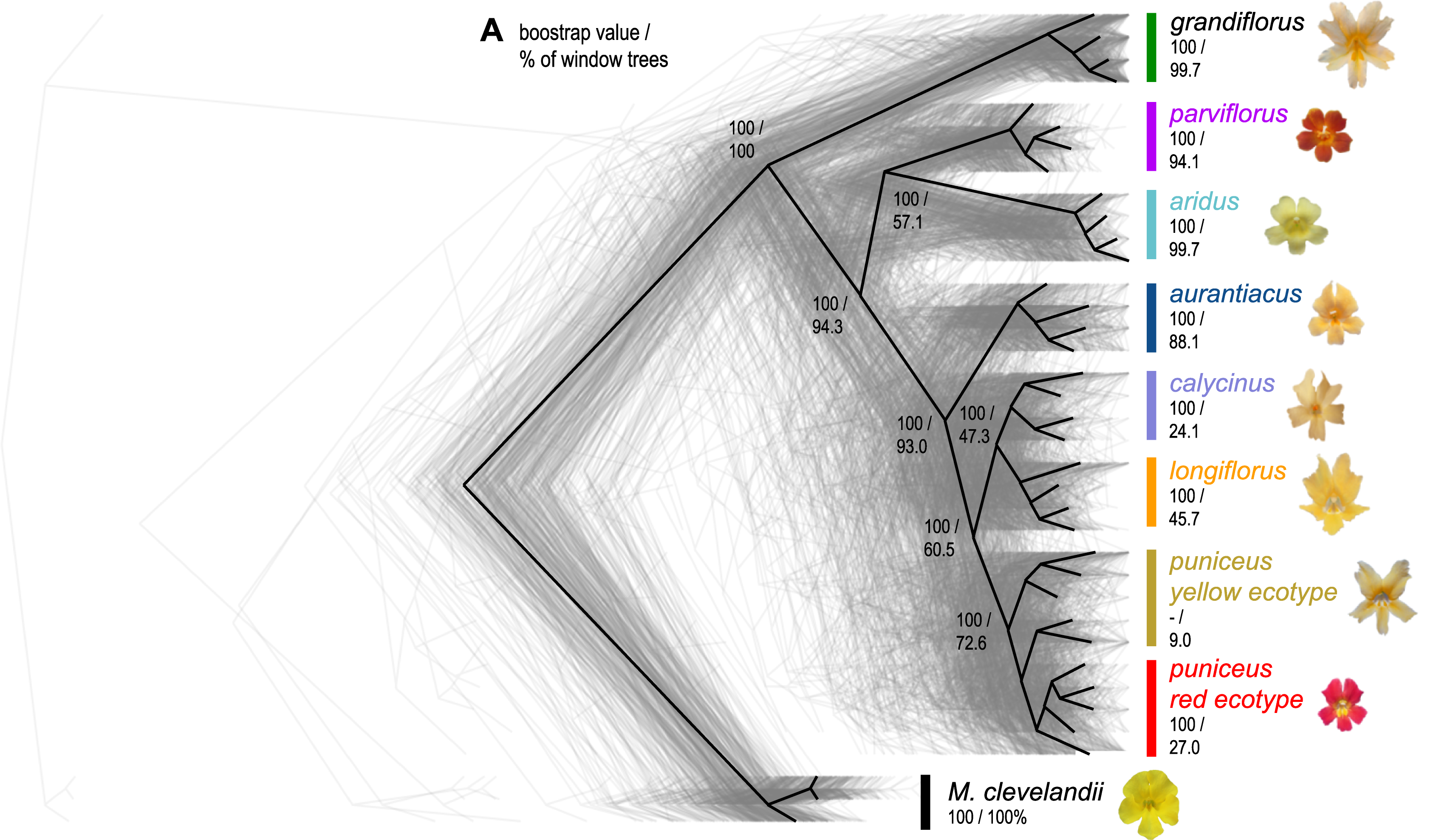
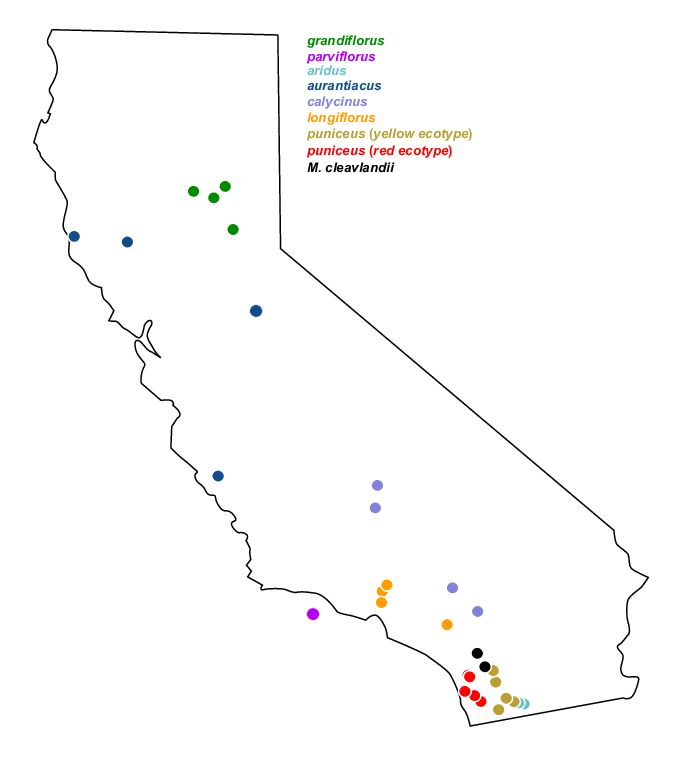
Some of the programs require we prepare population files. For pixy, this is a headerless, tab-separated file with sample and population columns. The sampleinfo.csv file contains the information we need; the sample names have been prefixed with a three-letter code to indicate population (apart from ssp. puniceus which also comes with a single letter R or Y indicating ecotype). Table 2 summarizes the populations and samples.
Code
# R code to generate sample population summary
data <- tibble(read.csv("sampleinfo.csv"))
data <- data %>%
mutate(Population = SampleAlias) %>%
mutate(across("Population", str_replace, "(.+)-.+$", "\\1")) %>%
mutate(across("Population", str_replace, "(CLV)_.+", "\\1"))
data %>%
group_by(Population, ScientificName) %>%
summarise(n = n(), samples = paste(SampleAlias, collapse = ", ")) %>%
kable()| Population | ScientificName | n | samples |
|---|---|---|---|
| ARI | Diplacus aridus | 4 | ARI-159_83, ARI-159_84, ARI-195_1, ARI-T84 |
| AUR | Diplacus aurantiacus | 4 | AUR-T102, AUR-T104, AUR-T50, AUR-T92 |
| CAL | Diplacus calycinus | 4 | CAL-T90, CAL-T91, CAL-T144, CAL-T150 |
| CLV | Diplacus clevelandii | 3 | CLV_GH, CLV_11, CLV_4 |
| GRA | Diplacus grandiflorus | 4 | GRA-T96, GRA-T101, GRA-T61, GRA-T99 |
| LON | Diplacus longiflorus | 4 | LON-T33, LON-T8, LON-DPR, LON-SS |
| PAR | Erythranthe parviflora | 4 | PAR-KK161, PAR-KK168, PAR-KK180, PAR-KK182 |
| PUN-R | Diplacus puniceus | 5 | PUN-R-ELF, PUN-R-JMC, PUN-R-LH, PUN-R-MT, PUN-R-UCSD |
| PUN-Y | Diplacus puniceus | 5 | PUN-Y-BCRD, PUN-Y-INJ, PUN-Y-LO, PUN-Y-PCT, PUN-Y-POTR |
We convert the sample information with csvtk. We also add a populations file with all samples belonging to the same population:
csvtk mutate --name Population --fields SampleAlias sampleinfo.csv |
csvtk cut --fields SampleAlias,Population |
csvtk replace --fields Population --pattern "(.+)-.+$" --replacement "\$1" |
csvtk replace --fields Population --pattern "(CLV)_.+" --replacement "\$1" |
csvtk del-header --out-tabs > populations.txt
csvtk cut --fields SampleAlias sampleinfo.csv |
csvtk mutate --name Population --fields SampleAlias |
csvtk replace --fields Population --pattern ".+" --replacement "ALL" |
csvtk del-header --out-tabs > populations.ALL.txtvcftools, on the other hand, requires that populations are specified as separate files, containing the individuals of each population. We can use the populations.txt file to quickly generate population-specific files, and we add an ALL population, treating all samples as coming from the same population:
for pop in ARI AUR CAL CLV GRA LON PAR PUN-R PUN-Y; do
csvtk --no-header-row grep --tabs --fields 2 --pattern "$pop" populations.txt | \
csvtk cut --tabs --fields 1 > $pop.txt;
done
csvtk cut --tabs --no-header-row --fields 1 populations.txt > ALL.txtWe define environment variables to make the downstream commands easier to type:
VCF=allsites.vcf.gz
POPS=populations.txtGenerating and visualising diversity statistics
Compiling statistics with vcftools
Create an output directory for the results and define some environment variables:
mkdir -p 01-vcftools
OUT=01-vcftools/allsitesNucleotide diversity (\(\pi\))
Nucleotide diversity can be calculated by site (--site-pi) or in windows (--window-pi)2.
vcftools --gzvcf $VCF --site-pi --out $OUT 2> /dev/null
csvtk summary $OUT.sites.pi --ignore-non-numbers --tabs \
--fields PI:mean,PI:stdevPI:mean PI:stdev
0.04 0.10# Set your window size higher, e.g., 10kb
vcftools --gzvcf $VCF --window-pi 1000 --out $OUT 2> /dev/null
On genome scans and window sizes
Genetic diversity estimates can be noisy, so we often want to compute values in sliding windows across a sequence. Choosing window size can be as simple as trying out different values, often ranging from single to several hundred kilobases. As always, the appropriate size depends on the analyses.
One rule of thumb that can be applied is that the window size should be larger than the genomic background of linkage disequilibrium (LD). Recall, LD is the non-random co-segregation of alleles at two or more loci. Linked loci will induce correlations in window-based statistics, so by choosing a window size larger than the LD background, we ensure that our windows are, in some sense, independent.
For this reason, a common approach is to calculate some measure of LD between marker pairs, and generate a plot of LD decay. This is outside the scope of this exercise, but the interested reader can consult the plink documentation for ways to do this. See also Figure 3 for an example plot.

Even though single point summary statistics can be informative, we can get an overview of the distribution over the chromosome by plotting:
csvtk plot line --tabs $OUT.windowed.pi -x BIN_START -y PI \
--point-size 0.01 --xlab "Position (bp)" \
--ylab "Diversity" --title LG4 --width 9.0 --height 3.5 \
> $OUT.png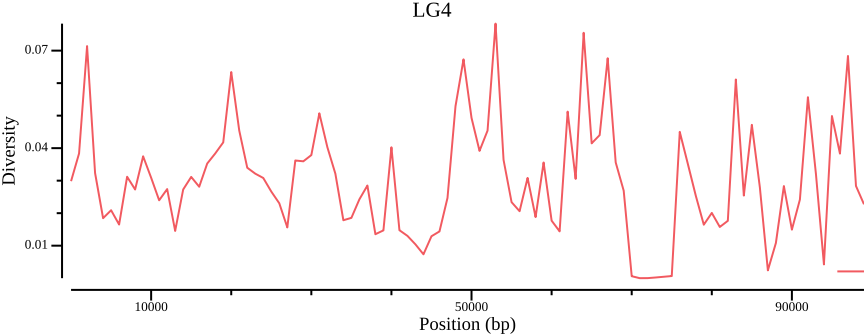
Figure 4 shows the diversity for all populations. We would also be interested in comparing the diversity for different populations. This can be achieved by passing a population file to bcftools view and piping (|) the output to vcftools:
bcftools view -S PAR.txt $VCF |\
vcftools --vcf - --window-pi 1000 --out $OUT.PAR 2> /dev/null
bcftools view -S ARI.txt $VCF |\
vcftools --vcf - --window-pi 1000 --out $OUT.ARI 2> /dev/nullWith some csvtk magic we can combine the measures and plot:
# When assigning a constant must enclose it in single quotes within double quotes
csvtk mutate2 --tabs --name Population --expression " 'ARI' " \
$OUT.ARI.windowed.pi > $OUT.ARI.wpi
csvtk mutate2 --tabs --name Population --expression " 'PAR' " \
$OUT.PAR.windowed.pi > $OUT.PAR.wpi
csvtk concat --tabs $OUT.ARI.wpi $OUT.PAR.wpi |\
csvtk plot --tabs line - -x BIN_START -y PI \
--group-field Population \
--point-size 0.01 --xlab "Position (bp)" \
--ylab "Diversity" --title "LG4:PAR and ARI" \
--width 9.0 --height 3.5 \
> $OUT.ARI.PAR.png 2>/dev/null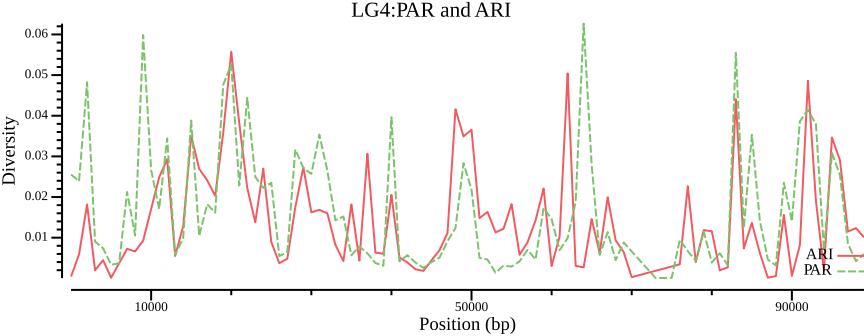
\(F_{ST}\)
Since \(F_{ST}\) is a statistic that compares populations, we must supply two or more of the population files we defined above. A population file name is passed to the --weir-fst-pop option. Calculations are done by site per default, but let’s calculate \(F_{ST}\) for a comparison of two populations in 100kb windows:
# Set your window size higher, e.g., 100000
vcftools --gzvcf $VCF --weir-fst-pop ARI.txt \
--weir-fst-pop PAR.txt \
--fst-window-size 1000 \
--out $OUT.ARI-PARcsvtk plot line --tabs $OUT.ARI-PAR.windowed.weir.fst \
-x BIN_START -y MEAN_FST \
--point-size 0.01 --xlab "Position (bp)" \
--ylab "Fst" --title "LG4:ARI vs PAR" \
--width 9.0 --height 3.5 \
> $OUT.ARI-PAR.windowed.weir.fst.mean.png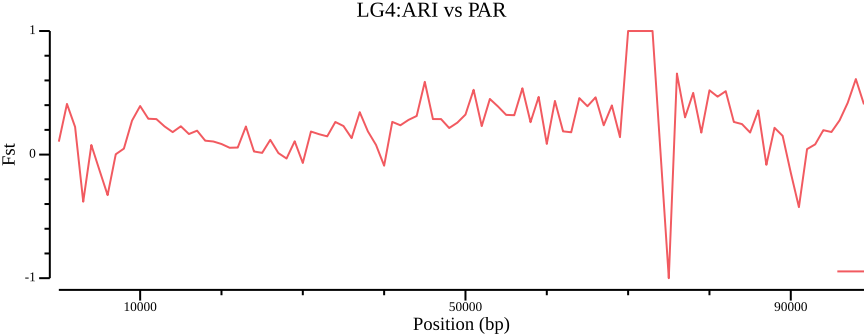
Compiling statistics with pixy
The vcftools statistics that we just calculated have a couple of issues:
- they have been calculated on unfiltered data; this can be somewhat remediated by applying a depth-based filter
- a more concerning problem is that even if we did apply a filter, all missing sites would be treated as invariant sites, when in reality the windows would need to be adjusted to the number of accessible sites within a window. This number could be calculated from a mask file, but adds some complexity to the calculations.
Instead of applying filters, let’s first try out an alternative solution. pixy is a program that facilitates the calculation of nucleotide diversity within \(\pi\) and between \(d_{XY}\) populations from a VCF, as well as differentiation (\(F_{ST}\)). It also takes invariant sites and missing data into consideration. Another nice feature is that by providing a population file with all samples, every population comparison is done on the fly, so only one run is needed!
Calculating per-site statistics takes too long time, so we will only generate windowed output here. This may still take a couple of minutes though.
mkdir -p 02-pixy
# Set your window size higher, e.g., 100kb
pixy --stats pi fst dxy \
--vcf $VCF \
--populations populations.txt \
--window_size 1000 \
--output_folder 02-pixy \
--n_cores 4head -n 3 02-pixy/pixy_pi.txtpop chromosome window_pos_1 window_pos_2 avg_pi no_sites count_diffs count_comparisons count_missing
LON LG4 1 1000 0.0019674355495251 616 29 14740 3544
PAR LG4 1 1000 0.0008201763379126 625 12 14631 3653For windowed output, the pixy output files contain information on the windows, the number of missing sites, number of snps, and more. The first column corresponds to the population, which means we can easily select lines matching population(s) of interest. For a full description, consult the documentation. Note that because we provided a population file defining two populations, diversity is calculated per population.
We conclude by plotting diversity for ARI and PAR
# Possibly remove NA values that otherwise would throw error
csvtk filter2 --tabs \
--filter '$avg_pi != "NA" && ($pop == "ARI" || $pop == "PAR") ' \
02-pixy/pixy_pi.txt | \
csvtk plot line --tabs - -x window_pos_1 -y avg_pi \
--group-field pop \
--point-size 0.01 --xlab "Position (bp)" \
--ylab "Diversity" --title "LG4:ARI and PAR" \
--width 9.0 --height 3.5 \
> 02-pixy/pixy_pi.ARI-PAR.txt.png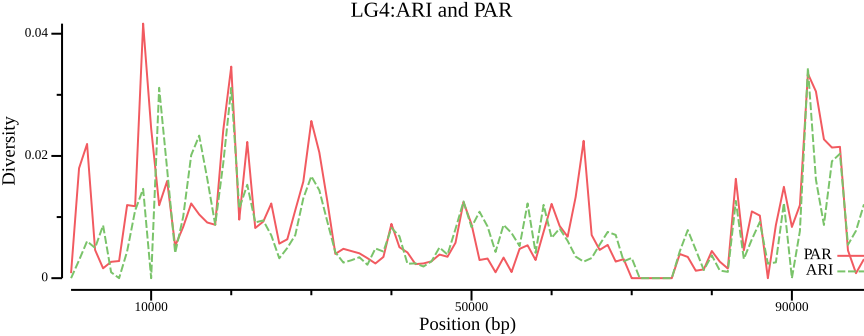
and \(F_{ST}\) comparing ARI and PAR
csvtk filter2 --tabs \
--filter '$avg_wc_fst != "NA" && $pop1 == "PAR" && $pop2 == "ARI" ' \
02-pixy/pixy_fst.txt | \
csvtk plot line -t - -x window_pos_1 -y avg_wc_fst \
--point-size 0.01 --xlab "Position (bp)" \
--ylab "Fst" --title "LG4:ARI vs PAR" --width 9.0 --height 3.5 \
> 02-pixy/pixy_fst.ARI-PAR.txt.png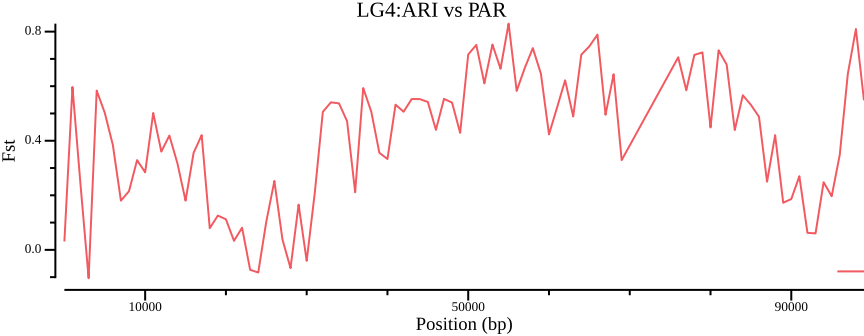
Comparing Figure 8 to Figure 6 shows a stark contrast, so clearly, how you filter and handle invariant/missing data will strongly affect the outcome.
Genome scans and genomic features
Working with Z-transformed data
Transforming raw data to Z-scores can facilitate the scan for outlier regions. However, there is no easy way to do this with csvtk, so here is some template R code to do this from pixy data.
pi <- read.table("pixy_pi.txt", header = TRUE)
# Select population PAR
data <- data[data$pop == "PAR", ]
x <- data$avg_pi
z <- (x - mean(x, na.rm = TRUE))/sd(x, na.rm = TRUE)
# Plot data along chromosome and identify region by eye plot(x =
# data$window_pos_1, y = z, xlab = 'Position (bp)')Stratifying by genomic feature
pixy accepts as input a BED file that defines coordinates of genome features. Here is an example of how to extract CDS regions and then compile results with pixy:
csvtk filter2 --tabs annotation.gff --filter ' $3 == "CDS" ' |\
csvtk cut --tabs --fields 1,10,11 | bedtools sort | bedtools merge \
> CDS.bed 2>/dev/nullpixy --vcf allsites.vcf.gz --stats pi \
--populations populations.txt \
--output_prefix cds --bed_file CDS.bedMonkeyflower diversity landscape
Drawing on what you learned earlier today about filtering, and with the help of the command examples above, you can now start exploring the Monkeyflower diversity landscape. Try different population comparisons, perform outlier analyses, and see if you can find candidate regions of interest. If you want inspiration, take a look at the methods section of Stankowski et al. (2019).
Here are some suggested actions that you can take, but feel free to look at the data in any way you want.
References
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., Handsaker, R. E., Lunter, G., Marth, G. T., Sherry, S. T., McVean, G., & Durbin, R. (2011). The variant call format and VCFtools. Bioinformatics (Oxford, England), 27(15), 2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., Whitwham, A., Keane, T., McCarthy, S. A., Davies, R. M., & Li, H. (2021). Twelve years of SAMtools and BCFtools. GigaScience, 10(2), giab008. https://doi.org/10.1093/gigascience/giab008
Fuller, Z. L., Mocellin, V. J. L., Morris, L. A., Cantin, N., Shepherd, J., Sarre, L., Peng, J., Liao, Y., Pickrell, J., Andolfatto, P., Matz, M., Bay, L. K., & Przeworski, M. (2020). Population genetics of the coral Acropora millepora: Toward genomic prediction of bleaching. Science, 369(6501), eaba4674. https://doi.org/10.1126/science.aba4674
Quinlan, A. R., & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26(6), 841–842. https://doi.org/10.1093/bioinformatics/btq033
Stankowski, S., Chase, M. A., Fuiten, A. M., Rodrigues, M. F., Ralph, P. L., & Streisfeld, M. A. (2019). Widespread selection and gene flow shape the genomic landscape during a radiation of monkeyflowers. PLOS Biology, 17(7), e3000391. https://doi.org/10.1371/journal.pbio.3000391