mkdir -p variant_calling
cd variant_callingVariant calling
Compute environment setup
If you haven’t already done so, please read Compute environment for information on how to prepare your working directory.
In this exercise we will produce a variant call set, going through the basic steps from quality control through read mapping to variant calling. We will be working on the Monkeyflowers dataset. Make sure to read the dataset document before running any commands as it will give you the biological background and general information about where to find and how to setup the data. We will focus on the red and yellow ecotypes in what follows.
Note
This is a technical exercise, where the focus is not so much on the biology as on getting programs to run and interpreting the output.
Important
The commands of this document have been run on a subset (a subregion) of the data. Therefore, although you will use the same commands, your results will differ from those presented here.
Learning objectives
- Perform qc on sequencing reads and interpret results
- Prepare reference for read mapping
- Map reads to reference
- Mark duplicates
- Perform raw variant calling to generate a set of sites to exclude from recalibration
- Perform base quality score recalibration
- Perform variant calling on base recalibrated data
- Do genotyping on all samples and combine results to a raw variant call set
Data setup
Move to your course working directory /proj/naiss2023-22-1084/users/USERNAME, create an exercise directory called monkeyflower and cd to it:
cd /proj/naiss2023-22-1084/users/USERNAME
mkdir -p monkeyflower
cd monkeyflowerThe variant calling exercise will be run on small datasets, so to avoid name clashes, create a directory variant_calling and cd to it:
Retrieve the data with rsync. You can use the --dry-run option to see what is going to happen; just remove it when you are content:
rsync --dry-run -av \
/proj/naiss2023-22-1084/webexport/monkeyflower/small/ .
# Remove the dry run option to actually retrieve data. The dot is
# important!
rsync -av /proj/naiss2023-22-1084/webexport/monkeyflower/small/ .Create an exercise directory called monkeyflower and cd to it:
mkdir -p monkeyflower
cd monkeyflowerThe variant calling exercise will be run on small datasets, so to avoid name clashes, create a directory variant_calling and cd to it:
mkdir -p variant_calling
cd variant_callingRetrieve the data https://export.uppmax.uu.se/naiss2023-22-1084/monkeyflower/small.tar.gz with wget1:
wget --user pgip --password PASSWORD https://export.uppmax.uu.se/naiss2023-22-1084/monkeyflower/small.tar.gz
tar -zxvf small.tar.gz
Tools
Execute the following command to load modules:
module load uppmax bioinfo-tools \
GATK/4.3.0.0 bwa/0.7.17 FastQC/0.11.9 \
MultiQC/1.12 picard/2.27.5 samtools/1.17 \
bcftools/1.17 QualiMap/2.2.1Copy the contents to a file environment.yml and install packages with mamba env update -f environment.yml.
channels:
- conda-forge
- bioconda
- default
dependencies:
- bcftools=1.15.1
- gatk4=4.4.0.0
- bwa=0.7.17
- fastqc=0.12.1
- multiqc=1.16
- picard=3.1.0
- qualimap=2.2.2d
- samtools=1.15.1Variant calling overview
A generic variant calling workflow consists of the following basic steps:
- read quality control and filtering
- read mapping
- removal / marking of duplicate reads
- joint / sample-based variant calling and genotyping
There are different tweaks and additions to each of these steps, depending on application and method.
1. Read quality control
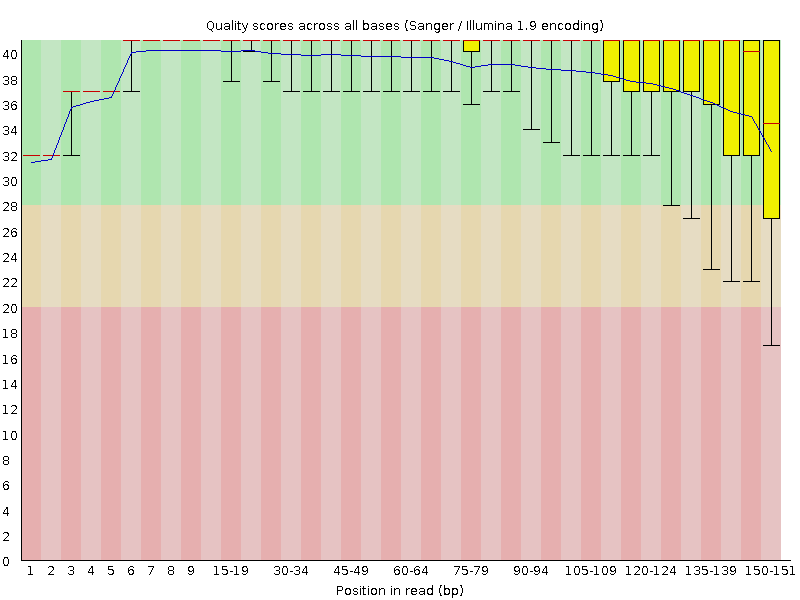
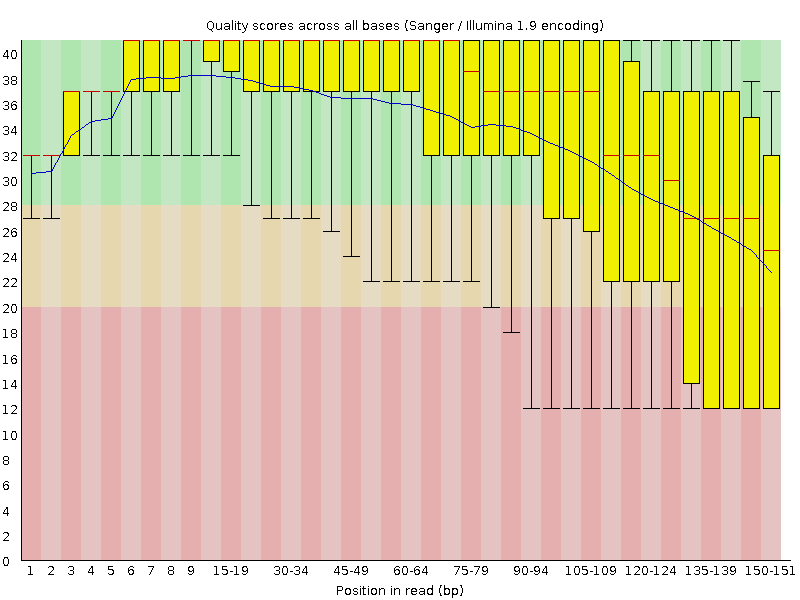
DNA sequencers score the quality of each sequenced base as phred quality scores, which is equivalent to the probability \(P\) that the call is incorrect. The base quality scores, denoted \(Q\), are defined as
\[ Q = -10 \log_{10} P \]
which for \(P=0.01\) gives \(Q=20\). Converting from quality to probability is done by solving for \(P\):
\[ P = 10^{-Q/10} \]
Hence, a base quality score \(Q=20\) (somtimes written Q20) corresponds to a 1% probability that the call is incorrect, Q30 a 0.1% probability, and so on, where the higher the quality score, the better. Bases with low quality scores are usually discarded from downstream analyses, but what is a good threshold? The human genome has approximately 1 SNP per 1,000 bp, which means sequencing errors will be ten times as probable in a single read for Q20 base calls. A reasonable threshold is therefore around Q20-Q30 for many purposes.
The base qualities typically drop towards the end of the reads (Figure 1). Prior to mapping it may therefore be prudent to remove reads that display too high drop in quality, too low mean quality, or on some other quality metric reported by the qc software.
The quality scores are encoded using ASCII codes. An example of a FASTQ sequence is given below. The code snippet shows an example of shell commands2 that are separated by a so-called pipe (|) character which takes the output from one process and sends it as input to the next3.
Note that we use the long option names to clarify commands, and we aim to do so consistently when a new command is introduced. Once you feel confident you know what a command does, you will probably want to switch to short option names, and we may do so in the instructions for some commonly used commands (e.g., head -n) without warning. Remember to use --help to examine command options.
# Command using long (-- prefix) option names
zcat PUN-Y-INJ_R1.fastq.gz | head --lines 4 | cut --characters -30
# Equivalent command using short (single -, single character) option names
# zcat PUN-Y-INJ_R1.fastq.gz | head -n 4 | cut -c -30@SRR9309790.10003134
TAAATCGATTCGTTTTTGCTATCTTCGTCT
+
AAFFFJJJJJJJFJJJJJJJJJJJJJJJJJA FASTQ entry consists of four lines:
- sequence id (prefixed by
@) - DNA sequence
- separator (
+) - phred base quality scores
2. Read mapping
Read mapping consists of aligning sequence reads, typically from individuals in a population (a.k.a. resequencing) to a reference sequence. The choice of read mapper depends, partly on preference, but mostly on the sequencing read length and application. For short reads, a common choice is bwa-mem, and for longer reads minimap2.
In what follows, we will assume that the sequencing protocol generates paired-end short reads (e.g., from Illumina). In practice, this means a DNA fragment has been sequenced from both ends, where fragment sizes have been selected such that reads do not overlap (i.e., there is unsequenced DNA between the reads of a given insert size).
The final output of read mapping is an alignment file in binary alignment map (BAM) format or variants thereof.
3. Removal / marking of duplicate reads
During sample preparation or DNA amplification with PCR, it may happen that a single DNA fragment is present in multiple copies and therefore produces redundant sequencing reads. This shows up as alignments with identical start and stop coordinates. These so-called duplicate reads should be marked prior to any downstream analyses. The most commonly used tools for this purpose are samtools markdup and picard MarkDuplicates.
4. Variant calling and genotyping
Once BAM files have been produced, it is time for variant calling, which is the process of identifying sites where there sequence variation. There are many different variant callers, of which we will mention four.
bcftools is a toolkit to process variant call files, but also has a variant caller command. We will use bcftools to look at and summarize the variant files.
freebayes uses a Bayesian model to call variants. It may be time-consuming in high-coverage regions, and one therefore may have to mask repetitive and other low-complexity regions.
ANGSD is optimized for low-coverage data. Genotypes aren’t called directly; rather, genotype likelihoods form the basis for all downstream analyses, such as calculation of diversity or other statistics.
Finally, GATK HaplotypeCaller performs local realignment around variant candidates, which avoids the need to run the legacy GATK IndelRealigner. Realignment improves results but requires more time to run. GATK is optimized for human data. For instance, performance drops dramatically if the reference sequence consists of many short scaffolds/contigs, and there is a size limit to how large the chromosomes can be. It also requires some parameter optimization and has a fairly complicated workflow (Hansen, 2016).
GATK best practice variant calling
We will base our work on the GATK Germline short variant discovery workflow. In addition to the steps outlined above, there is a step where quality scores are recalibrated in an attempt to correct errors produced by the base calling procedure itself.
GATK comes with a large set of tools. For a complete list and documentation, see the “Tool Documentation Index” (2023).
Preparation: reference sequence index and read QC
Prior to mapping we need to create a database index. We also generate a fasta index and a sequence dictionary for use with the picard toolkit.
Important
On UPPMAX, the command to run picard is java -jar $PICARD_HOME/picard.jar. The UPPMAX command version is included as a comment whenever there is a command running picard. Make sure to uncomment when copying the code. If you have problems running the command, it may be because an incompatible java version is loaded as a module. To remedy this situation, run
module unload javasamtools faidx M_aurantiacus_v1.fasta
# java -jar $PICARD_HOME/picard.jar CreateSequenceDictionary --REFERENCE M_aurantiacus_v1.fasta
picard CreateSequenceDictionary --REFERENCE M_aurantiacus_v1.fasta
bwa index M_aurantiacus_v1.fastaWith the program fastqc we can generate quality control reports for all input FASTQ files simultaneously, setting the output directory with the -o flag:
# Make fastqc output directory; --parents makes parent directories as
# needed
mkdir --parents fastqc
fastqc --outdir fastqc *fastq.gzWe will use MultiQC later on to combine the results from several output reports.
Read mapping
We will start by mapping FASTQ read pairs to the reference. We will use the bwa read mapper together with samtools to process the resulting output.
Read group information identifies sequence sets
Some of the downstream processes require that reads have been assigned read groups (“Read Groups,” 2023), which is a compact string representation of a set of reads that originate from a sequencing unit and sample. Assigning read groups becomes particularly important for multiplexing protocols, or when a sample has been sequenced on different lanes or platform units, as it allows for the identification of sequence batch issues (e.g., poor sequence quality). Here, we want to assign a unique ID, the sample id (SM), and the sequencing platform (PL), where the latter informs the algorithms on what error model to apply. The read group is formatted as @RG\tID:uniqueid\tSM:sampleid\tPU:platform, where \t is the tab character. More fields can be added; see the SAM specification, section 1.3 (HTS Format Specifications, 2023) for a complete list.
Sample information is available in the sampleinfo.csv file:
head -n 3 sampleinfo.csvSample,Run,ScientificName,SampleName,AuthorSample,SampleAlias,Taxon,Latitude,Longitude,% Reads aligned,Seq. Depth
SRS4979271,SRR9309782,Diplacus longiflorus,LON-T33_1,T33,LON-T33,ssp. longiflorus,34.3438,-118.5099,94.6,18.87
SRS4979267,SRR9309785,Diplacus longiflorus,LON-T8_8,T8,LON-T8,ssp. longiflorus,34.1347,-118.6452,82.6,25.11The sample information is a combination of the run information obtained from the SRA (BioProject PRJNA549183) and the sample sheet provided with the article. An additional column SampleAlias has been added that names samples using a three-letter abbreviation for population hyphenated with the sample identifier. For the ssp. puniceus, an additional one-letter character denoting the color ecotype is inserted between population and sample id. PUN-Y-BCRD then is a sample from the puniceus subspecies with the yellow ecotype. We will use the Run column as unique ID, SampleAlias as the sample id SM, and ILLUMINA as the platform PL.
Read mapping with bwa and conversion to BAM format with samtools
Let’s map the FASTQ files corresponding to sample PUN-Y-BCRD:
bwa mem -R "@RG\tID:SRR9309788\tSM:PUN-Y-BCRD\tPL:ILLUMINA" -t 4 \
-M M_aurantiacus_v1.fasta PUN-Y-BCRD_R1.fastq.gz PUN-Y-BCRD_R2.fastq.gz | \
samtools sort - | samtools view --with-header --output PUN-Y-BCRD.sort.bamThere’s a lot to unpack here. First, the -R flag to bwa mem passes the read group information to the mapper. -t sets the number of threads, and -M marks shorter split hits as secondary, which is for Picard compatibility4. The first positional argument is the reference sequence, followed by the FASTQ files for read 1 and 2, respectively.
The output would be printed on the screen (try running the bwa mem command alone!), but we pipe the output to samtools sort to sort the mappings (by default by coordinate). The - simply means “read from the pipe”.
Finally, samtools view converts the text output to binary format (default), including the header information (short option -h). You can use the same command to view the resulting output on your screen:
samtools view PUN-Y-BCRD.sort.bam | head -n 2SRR9309788.7313829 129 LG4 29 60 103M2I46M = 83824 83796 GTCAATTTCATGTTTGACTTTTAGATTTTTAATTAATTATATATTTTTTGCAATTTGTAACCTCTTTAACCTTTATTTAATTTTTTGAATTTCTTTTTTATTTTATTTTCAAATACAATTCACCCCAATTAATTATTTTAATTATAACAAT AAFFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJAFJJFJJJF<JJJJJJJJ<AJJJJJJJJJ-FFJJAJJJJA-7-7----<A---7<-)7AAA-AA7-<-7--<A---7---7 NM:i:6 MD:Z:107T25A1C6A6 MC:Z:88M8D63M AS:i:121 XS:i:80 RG:Z:SRR9309788
SRR9309788.9554822 99 LG4 58 60 74M2I75M = 256 321 TAATTAATTATATATTTTTTGCAATTTGTAACCTCTTTAACCTTTATTTAATTTTTTGAATTTCTTTTTTATTTTATTTTCAAATACAATTCACCCCAATTAATTAATCTAATTAAAACAATTAAATAATCAACCCGAATGATTAACCAAT AAFFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFAAJJJJJJJJFJJJFJJ<FJJAJJJFJJJJ< NM:i:5 MD:Z:78T54G0C14 MC:Z:21S82M2I9M5I32M AS:i:126 XS:i:81 RG:Z:SRR9309788Mapping from uBAM file
There is an alternative storage format for the FASTQ files called uBAM (unmapped BAM format). The GATK developers promote its use in lieu of FASTQ files since BAM files can store more metadata associated with sequencing runs. The workflow is slightly more complex, but the files have been prepared for you so you don’t need to worry about generating the uBAM files.
To facilitate downstream processing, we will from now on make use of environment variables5 to refer to a sample and the reference sequence. Retrieve the SRR id from the sampleinfo file.
export SRR=SRR9309790
export SAMPLE=PUN-Y-INJ
export REF=M_aurantiacus_v1.fasta
samtools fastq ${SAMPLE}.unmapped.bam | \
bwa mem -R "@RG\tID:${SRR}\tSM:${SAMPLE}\tPL:ILLUMINA" -t 4 -p -M ${REF} - | \
samtools sort - | samtools view -h -o ${SAMPLE}.sort.bamNote that we here need to use the command samtools fastq to read the contents of ${SAMPLE}.unmapped.bam and pipe the output to bwa mem.
Mark duplicate reads with Picard MarkDuplicates
Once mapping is completed, we must find and mark duplicate reads as these can distort the results of downstream analyses, such as variant calling. We here use Picard MarkDuplicates6:
#java -jar $PICARD_HOME/picard.jar MarkDuplicates --INPUT ${SAMPLE}.sort.bam \
# --METRICS_FILE ${SAMPLE}.sort.dup_metrics.txt \
# --OUTPUT ${SAMPLE}.sort.dup.bam
picard MarkDuplicates --INPUT ${SAMPLE}.sort.bam \
--METRICS_FILE ${SAMPLE}.sort.dup_metrics.txt \
--OUTPUT ${SAMPLE}.sort.dup.bamThe metrics output file contains information on the rate of duplication. We will include the output in the final MultiQC report.
An additional mapping quality metric of interest is percentage mapped reads and average read depth. We can use qualimap bamqc to collect mapping statistics from a BAM file:
qualimap bamqc -bam ${SAMPLE}.sort.bamA summary of the results is exported to ${SAMPLE}.sort_stats/genome_results.txt; we show percent mapping and average coverage below as examples:
grep "number of mapped reads" ${SAMPLE}.sort_stats/genome_results.txt
grep "mean coverageData" ${SAMPLE}.sort_stats/genome_results.txt number of mapped reads = 7,643 (96.94%)
mean coverageData = 11.0246X
Why does the sample have such low coverage?
If you look at the coverage reported in sampleinfo.csv, column Seq. Depth, you will note that the observed coverage here is much lower. This has to do with how the exercise data set was prepared; only read pairs where both reads mapped within the example region were retained.
These statistics will also be picked up by MultiQC.
Generate high quality known sites for BQSR
Once we have a duplicate marked BAM file, we can proceed with Base Quality Score Recalibration (BQSR). The purpose here is to correct any systematic errors made by the sequencing machine. This is done by applying a machine learning model. Before we do so, however, we need to generate a list of known sites that should not be recalibrated7 Therefore, we will first perform a preliminary round of variant calling and filtering to generate a known sites callset.
gatk HaplotypeCaller -OVI true \
--emit-ref-confidence GVCF \
--annotation FisherStrand -A QualByDepth -A MappingQuality -G StandardAnnotation \
--input ${SAMPLE}.sort.dup.bam --output ${SAMPLE}.sort.dup.raw.hc.g.vcf.gz \
--reference ${REF}
gatk VariantFiltration -OVI true \
--variant ${SAMPLE}.sort.dup.raw.hc.g.vcf.gz \
--output ${SAMPLE}.sort.dup.raw.hc.filtered.g.vcf.gz \
--filter-name FisherStrand --filter 'FS > 50.0' \
--filter-name QualByDepth --filter 'QD < 4.0' \
--filter-name MappingQuality --filter 'MQ < 50.0'HaplotypeCaller is GATK’s variant caller that calls both SNPs and indels by realigning sequences in the vicinity of sites that harbor variation. We apply the --emit-ref-confidence (-ERC) option to generate GVCF output format, a condensed VCF format that includes non-variant sites as well as variant sites. The --annotation (-A) option adds annotations to the output that are used in the subsequent filtering step.
VariantFiltration does what its name suggests. The --filter-name / --filter option pairs apply named filters to the input data. For instance, we here remove variants whose mapping quality is below 50.0.
Now we have per-sample known sites callsets that can be used as input to BQSR.
Base quality score recalibration
Base quality score recalibration consists of two commands:
gatk BaseRecalibrator --input ${SAMPLE}.sort.dup.bam \
--reference ${REF} --known-sites ${SAMPLE}.sort.dup.raw.hc.filtered.g.vcf.gz \
--output ${SAMPLE}.sort.dup.recal.table
gatk ApplyBQSR --bqsr-recal-file ${SAMPLE}.sort.dup.recal.table \
--input ${SAMPLE}.sort.dup.bam --output ${SAMPLE}.sort.dup.recal.bamBaseRecalibrator compiles empirical data for all non-variant sites for an input BAM file for a number of covariates (e.g., nucleotide context). The basic idea is to fit a probability of error (i.e., a quality score of sorts), to observed covariate combinations, which is recorded in a recalibration table file. The probability is then used correct the quality scores reported by the sequencing machine in ApplyBQSR.
OPTIONAL: Examine the output of BQSR
We can investigate how well BQSR has performed by applying two more commands. First, we generate a calibration table for the recalibrated BAM file:
gatk BaseRecalibrator --input ${SAMPLE}.sort.dup.recal.bam \
--reference ${REF} --known-sites ${SAMPLE}.sort.dup.raw.hc.filtered.g.vcf.gz \
--output ${SAMPLE}.sort.dup.recal.after.tableThen we use the two calibration tables as input to AnalyzeCovariates to generate a pdf output (and a csv file that we will use in the next code block):
gatk AnalyzeCovariates --before-report-file ${SAMPLE}.sort.dup.recal.table \
--after-report-file ${SAMPLE}.sort.dup.recal.after.table \
--plots-report-file ${SAMPLE}.sort.dup.recal.after.pdf \
--intermediate-csv-file ${SAMPLE}.sort.dup.recal.after.csvFigure 2 shows an example plot of quality values for base substitutions. There are more plots and summaries in the pdf output file.

Several rounds of generating known sites followed by BQSR can be applied until convergence. This means that running BQSR can be time-consuming, and one side effect is that the resulting BAM output files can become very large, which may or not be a problem. Also, sequencing technologies keep improving, potentially eliminating concerns of biased quality scores. This begs the question: is it worth running BQSR? For the time being, the GATK developers do recommend that BQSR always be run. Keep in mind though, that for non-model organisms, one issue is that there seldom is a catalogue of known sites, which means the user has to bootstrap such a call set, like we did above. For a more complete discussion, see the technical documentation on BQSR.
Variant calling and genotyping
After we have generated the recalibrated BAM files, we can proceed with the “real” variant calling. We once again run GATK HaplotypeCaller and VariantFiltration.
That concludes the sample-specific part of the variant calling workflow! Now we need to combine the samples and perform genotyping. To do so, we need to run the same commands, from mapping to variant calling and filtering on recalibrated files, for all samples.
Compile QC metrics
Identifying sample quality issues is crucial for downstream processing. Quality metrics that indicate problems with a sample could potentially lead to its removal entirely from subsequent analyses. Many programs generate QC metrics, but as it is difficult to get an overview by sifting through all separate QC reports, it is recommended that you compile them with MultiQC. Running MultiQC is as simple as
multiqc .Open the report multiqc_report.html to quickly QC your data. There are lots of ways you can interactively modify the report to make more logical groupings (e.g., grouping reports by sample9) , but we won’t go into this topic in more detail here.
Combine genomic VCF files and genotype
We are now at the stage where we can do genotyping of our samples. This done by first combining individual sample gVCF files with GATK CombineGVCFs, followed by genotyping with GenotypeGVCFs. We use the label subset to indicate that we are looking at a sample subset.
# -OVI = --create-output-variant-index
# -R = --reference
gatk CombineGVCFs -OVI true --output combine.subset.g.vcf.gz --reference ${REF} \
--variant PUN-Y-INJ.sort.dup.recal.hc.g.vcf.gz \
--variant PUN-R-ELF.sort.dup.recal.hc.g.vcf.gz
gatk GenotypeGVCFs -OVI true -R ${REF} -V combine.subset.g.vcf.gz \
--output variantsites.subset.vcf.gz
gatk GenotypeGVCFs -OVI true -R ${REF} -V combine.subset.g.vcf.gz \
--output allsites.subset.vcf.gz --all-sitesHere, the option --all-sites tells gatk GenotypeGVCFs to include variant as well as invariant sites in the output. Many software packages that are based on genetic diversity statistics implicitly assume that invariant sites are all homozygous reference (Korunes & Samuk, 2021). However, if invariant sites lack sequencing coverage, they should preferably be treated as missing data, which theoretically could harbour (unobserved) variant sites. Therefore, the inclusion of invariant sites provides a way to generate estimates of missing data, based on, e.g., sequencing depth profiles.
Inclusion of invariant sites comes at a cost however; the VCF file size may increase dramatically, to the point that it becomes impractical or even impossible to process. We generate both types of files for future analyses.
Looking closer at variants with bcftools
As a final step, let’s run a couple of commands to view variants and summarize variant statistics.
Compiling statistics
First out is bcftools stats, which compiles a summary of statistics, such as number of variants, quality distribution over variants, indel distributions and more.
bcftools stats allsites.vcf.gz > allsites.vcf.gz.statsThe output text file consists of report sections that can be easily extracted with the grep command by virtue of the fact that the first column corresponds to a shortcode of the statistic. For instance, SN corresponds to Summary numbers:
grep "^SN" allsites.vcf.gz.statsSN 0 number of samples: 10
SN 0 number of records: 100000
SN 0 number of no-ALTs: 91890
SN 0 number of SNPs: 3980
SN 0 number of MNPs: 0
SN 0 number of indels: 1197
SN 0 number of others: 0
SN 0 number of multiallelic sites: 541
SN 0 number of multiallelic SNP sites: 69Here, we can see that we have included non-variant sites (no-ALTs), that there are indels, and that a number of sites are multiallelic (more than two alleles).
Rerunning MultiQC will automatically add this report.
# Use --force (-f) to overwrite old report
multiqc --force .We will use bcftools stats in the next exercise to look more closely at variant quality metrics.
Looking inside the VCF
A Variant Call Format (VCF) file consists of three sections: meta-information lines (prefixed with ##), a header line (prefixed with #), followed by the data. The meta-information contains provenance information detailing how the file was generated, FILTER specification, INFO fields that provide additional information to genotypes, FORMAT specification fields that define genotype entries, and more. You can print the header10 with the command bcftools view -h. An example of each is given below:
bcftools view --header-only allsites.vcf.gz | grep "##FILTER" | head -n 1
bcftools view -h allsites.vcf.gz | grep "##INFO" | head -n 1
bcftools view -h allsites.vcf.gz | grep "##FORMAT" | head -n 1##FILTER=<ID=PASS,Description="All filters passed">
##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele count in genotypes, for each ALT allele, in the same order as listed">
##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Allelic depths for the ref and alt alleles in the order listed">The header field consists of eight mandatory columns CHROM, POS, ID, REF, ALT, QUAL, FILTER and INFO, followed by FORMAT and sample columns that contain the called genotypes:
bcftools view -h allsites.vcf.gz | grep CHROM#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT PUN-R-ELF PUN-R-JMC PUN-R-LH PUN-R-MT PUN-R-UCSD PUN-Y-BCRD PUN-Y-INJ PUN-Y-LO PUN-Y-PCT PUN-Y-POTRLet’s look at the first SNP entry (here, the first line extracts the position of the SNP):
pos=$(bcftools view --types snps --samples PUN-R-JMC allsites.vcf.gz \
--no-header | head -n 1 | cut -f 2)
bcftools view -v snps -s PUN-R-JMC allsites.vcf.gz LG4:$pos | tail -n 2#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT PUN-R-JMC
LG4 17 . C T 368.55 . AC=0;AF=0.429;AN=0;DP=22;ExcessHet=0;FS=0;InbreedingCoeff=0.5794;MLEAC=7;MLEAF=0.5;MQ=55.94;QD=33.5;SOR=0.859 GT:AD:DP:GQ:PGT:PID:PL:PS ./.:1,0:1:0:.:.:0,0,0:.The most important columns are CHROM, which indicates the sequence (LG4), the genome position POS, the reference allele REF (i.e., the nucleotide in the reference sequence), and the alternate allele ALT, which is the called variant. QUAL is a Phred-scaled quality of the call and can be used to filter low-quality calls. The FILTER column can be used to set filter flags. The INFO column contains metadata concerning the site; for instance, DP is the approximate read depth, the definition of which is included in the meta-information:
bcftools view -h allsites.vcf.gz | grep INFO | grep "ID=DP"##INFO=<ID=DP,Number=1,Type=Integer,Description="Approximate read depth; some reads may have been filtered">Finally, the columns following FORMAT pertain to the samples and contain the genotype calls, formatted according to the - you guessed it - FORMAT column. The example above contains a number of fields, where GT is the genotype:
bcftools view -h allsites.vcf.gz | grep FORMAT | grep "ID=GT"##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">For diploid samples, the genotype format is #/#, or #|# for phased data, where the hash marks are numbers that refer to the reference (0) and alternate (1 or higher) alleles. A . indicates a missing call.
Viewing variants and regions
The bcftools view command allows for quick access and viewing of file contents. A prerequisite is that we first index the file:
bcftools index allsites.vcf.gzafter which we can view, say, the first three indel variants in the region 12,010,000-12,010,100, corresponding to VCF coordinates 10,000-10,10011:
bcftools view -v indels -H allsites.vcf.gz LG4:10000-10100LG4 10048 . T TC 723.24 . AC=5;AF=0.278;AN=18;BaseQRankSum=2.17;DP=79;ExcessHet=0.3476;FS=9.691;InbreedingCoeff=0.1176;MLEAC=6;MLEAF=0.333;MQ=60;MQRankSum=0;QD=18.08;ReadPosRankSum=1.1;SOR=1.502 GT:AD:DP:GQ:PGT:PID:PL:PS 0|1:6,4:10:99:0|1:10048_T_TC:104,0,240:10048 0/0:5,0:5:15:.:.:0,15,149:. 0/0:11,0:11:33:.:.:0,33,299:. 0/1:6,6:12:99:.:.:166,0,167:. 1/1:0,8:8:24:.:.:269,24,0:. 0/1:3,7:10:99:.:.:203,0,105:. 0/0:6,0:6:18:.:.:0,18,173:. ./.:0,0:0:0:.:.:0,0,0:. 0/0:7,0:7:0:.:.:0,0,144:. 0/0:10,0:10:30:.:.:0,30,288:.bcftools view has many options for subsetting and filtering variants. Remember to consult the help pages!
Selecting samples
Finally, we look at how we can select samples from a variant file. To list the samples in a file, we can run bcftools query:
bcftools query --list-samples allsites.vcf.gzPUN-R-ELF
PUN-R-JMC
PUN-R-LH
PUN-R-MT
PUN-R-UCSD
PUN-Y-BCRD
PUN-Y-INJ
PUN-Y-LO
PUN-Y-PCT
PUN-Y-POTRSelecting indel variants for a given sample, say PUN-Y-BCRD, and region LG4:12001000-12001100 can then be done as follows:
bcftools view -H -s PUN-Y-BCRD -v indels allsites.vcf.gz LG4:1000-1100LG4 1000 . A AC 2143.22 . AC=2;AF=0.778;AN=2;DP=60;ExcessHet=0;FS=0;InbreedingCoeff=0.5649;MLEAC=16;MLEAF=0.889;MQ=60;QD=27.65;SOR=1.524 GT:AD:DP:GQ:PL 1/1:0,8:8:24:323,24,0
LG4 1023 . AAT A 2337.16 . AC=2;AF=1;AN=2;DP=59;ExcessHet=0;FS=0;InbreedingCoeff=0.588;MLEAC=17;MLEAF=1;MQ=60;QD=29.48;SOR=1.094 GT:AD:DP:GQ:PGT:PID:PL:PS 1|1:0,7:7:21:1|1:1023_AAT_A:315,21,0:1023
LG4 1040 . TG T 2337.04 . AC=2;AF=1;AN=2;DP=56;ExcessHet=0;FS=0;InbreedingCoeff=0.5807;MLEAC=17;MLEAF=1;MQ=60;QD=30.56;SOR=1.094 GT:AD:DP:GQ:PGT:PID:PL:PS 1/1:0,7:7:21:.:.:315,21,0:.
LG4 1047 . AG A 2246.22 . AC=2;AF=1;AN=2;DP=54;ExcessHet=0;FS=0;InbreedingCoeff=0.5664;MLEAC=17;MLEAF=1;MQ=60;QD=30.55;SOR=1.007 GT:AD:DP:GQ:PGT:PID:PL:PS 1|1:0,7:7:21:1|1:1023_AAT_A:315,21,0:1023
LG4 1053 . AG A 2201.19 . AC=2;AF=1;AN=2;DP=53;ExcessHet=0;FS=0;InbreedingCoeff=0.5659;MLEAC=17;MLEAF=1;MQ=60;QD=32.62;SOR=0.963 GT:AD:DP:GQ:PGT:PID:PL:PS 1|1:0,7:7:21:1|1:1023_AAT_A:315,21,0:1023References
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., Whitwham, A., Keane, T., McCarthy, S. A., Davies, R. M., & Li, H. (2021). Twelve years of SAMtools and BCFtools. GigaScience, 10(2), giab008. https://doi.org/10.1093/gigascience/giab008
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., Philippakis, A. A., del Angel, G., Rivas, M. A., Hanna, M., McKenna, A., Fennell, T. J., Kernytsky, A. M., Sivachenko, A. Y., Cibulskis, K., Gabriel, S. B., Altshuler, D., & Daly, M. J. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics, 43(5), 491–498. https://doi.org/10.1038/ng.806
Ewels, P., Magnusson, M., Lundin, S., & Käller, M. (2016). MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32(19), 3047–3048. https://doi.org/10.1093/bioinformatics/btw354
Hansen, N. F. (2016). Variant Calling From Next Generation Sequence Data. In E. Mathé & S. Davis (Eds.), Statistical Genomics: Methods and Protocols (pp. 209–224). Springer. https://doi.org/10.1007/978-1-4939-3578-9_11
HTS format specifications. (2023). https://samtools.github.io/hts-specs/
Korunes, K. L., & Samuk, K. (2021). Pixy: Unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Molecular Ecology Resources, 21(4), 1359–1368. https://doi.org/10.1111/1755-0998.13326
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-Bio]. https://arxiv.org/abs/1303.3997
Okonechnikov, K., Conesa, A., & García-Alcalde, F. (2016). Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics, 32(2), 292–294. https://doi.org/10.1093/bioinformatics/btv566
Picard toolkit. (2019). In Broad Institute, GitHub repository. https://broadinstitute.github.io/picard/; Broad Institute.
Read groups. (2023). In GATK. https://gatk.broadinstitute.org/hc/en-us/articles/360035890671-Read-groups
Tool Documentation Index. (2023). In GATK. https://gatk.broadinstitute.org/hc/en-us/articles/13832655155099--Tool-Documentation-Index
Footnotes
The password is provided by the course instructor↩︎
For any shell command, use the option
--helpto print information about the commands and its options.zcatis a variant of thecatcommand that prints the contents of a file on the terminal; thezprefix shows the command works on compressed files, a common naming convention.headviews the first lines of a file, andcutcan be used to cut out columns from a tab-delimited file, or in this case, cut the longest strings to 30 characters width.↩︎For more information, see unix pipelines↩︎
bwaconsistently uses short option names. Also, there is no--helpoption. To get a list of options, at the command line simply typebwa mem, orman bwa memfor general help and a complete list of options.↩︎Briefly, environment variables are a great way to generalise commands. To reuse the command, one only needs to modify the value of the variable.↩︎
The actual command call will depend on how Picard was installed. The conda installation provides access via the
picardwrapper, whereas on UPPMAX you must pointjavato the actual jar file (java -jar $PICARD_ROOT/picard.jar)↩︎If we don’t supply a list of known sites, the variants will be treated as errors and therefore recalibrated to the reference state.↩︎
Some programs, such as GATK, print their help output to
stderr, one of two so-called standard output streams (the other beingstdout). The2>&1is a construct that redirects (>) the output from stderr (2) to stdout (1) and does so in the background (&). By piping tolesswe can scroll through the documentation.↩︎MultiQC does a fairly good job at this as it is, but it does so by guessing sample names from file names. You may need to help it along the way by using the rename tab.↩︎
Which in this context means both the metadata information and header line↩︎
Although the reference sequence corresponds to region LG4:12000000-12100000, the coordinates in
allsites.vcf.gzstart from 1↩︎