cd /home/nikolay/Documents/Teaching/PopGen_2023
mkdir population_structure
cd population_structure
pwd/home/nikolay/Documents/Teaching/PopGen_2023/population_structure
In this notebook, we will apply PCA and Admixture analyses to real human data from the 1000 genomes project that aims to explore genetic diversity of human populations across the globe.
We will be using low-coverage Phase 1 human from from the 1000G project. The data are not heavy, so easy to download and work with, and one can test a few popgen methods using these real data. First, you will need to make a directory, for example population_structure, for running the population structure analysis.
In [1]:
cd /home/nikolay/Documents/Teaching/PopGen_2023
mkdir population_structure
cd population_structure
pwd/home/nikolay/Documents/Teaching/PopGen_2023/population_structure
Next, we will download the bam-alignments from the 1000G project and unzip them.
In [2]:
wget https://export.uppmax.uu.se/uppstore2018095/1000G.tar.gz
#wget https://export.uppmax.uu.se/naiss2023-22-1084/1000G.tar.gz
tar -xzf 1000G.tar.gz
rm 1000G.tar.gz--2023-11-07 13:14:16-- https://export.uppmax.uu.se/uppstore2018095/1000G.tar.gz
Resolving export.uppmax.uu.se (export.uppmax.uu.se)... 89.44.248.44
Connecting to export.uppmax.uu.se (export.uppmax.uu.se)|89.44.248.44|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 294605223 (281M) [application/x-gzip]
Saving to: ‘1000G.tar.gz’
1000G.tar.gz 0%[ ] 0 --.-KB/s 1000G.tar.gz 1%[ ] 2,95M 14,7MB/s 1000G.tar.gz 2%[ ] 8,27M 20,7MB/s 1000G.tar.gz 4%[ ] 13,65M 22,7MB/s 1000G.tar.gz 6%[> ] 19,05M 23,8MB/s 1000G.tar.gz 9%[> ] 26,20M 26,2MB/s 1000G.tar.gz 11%[=> ] 33,34M 27,8MB/s 1000G.tar.gz 14%[=> ] 40,50M 28,9MB/s 1000G.tar.gz 16%[==> ] 47,65M 29,8MB/s 1000G.tar.gz 19%[==> ] 54,78M 30,4MB/s 1000G.tar.gz 22%[===> ] 61,96M 31,0MB/s 1000G.tar.gz 24%[===> ] 69,09M 31,4MB/s 1000G.tar.gz 27%[====> ] 76,45M 31,8MB/s 1000G.tar.gz 29%[====> ] 83,61M 32,1MB/s 1000G.tar.gz 32%[=====> ] 91,00M 32,5MB/s 1000G.tar.gz 34%[=====> ] 98,15M 32,7MB/s eta 6s 1000G.tar.gz 37%[======> ] 105,34M 33,8MB/s eta 6s 1000G.tar.gz 40%[=======> ] 112,47M 34,4MB/s eta 6s 1000G.tar.gz 42%[=======> ] 119,63M 35,3MB/s eta 6s 1000G.tar.gz 45%[========> ] 126,79M 35,7MB/s eta 6s 1000G.tar.gz 47%[========> ] 133,95M 35,9MB/s eta 4s 1000G.tar.gz 50%[=========> ] 141,10M 35,9MB/s eta 4s 1000G.tar.gz 52%[=========> ] 147,97M 35,8MB/s eta 4s 1000G.tar.gz 55%[==========> ] 155,16M 35,8MB/s eta 4s 1000G.tar.gz 57%[==========> ] 162,31M 35,8MB/s eta 4s 1000G.tar.gz 60%[===========> ] 169,42M 35,8MB/s eta 3s 1000G.tar.gz 62%[===========> ] 176,60M 35,8MB/s eta 3s 1000G.tar.gz 65%[============> ] 183,77M 35,8MB/s eta 3s 1000G.tar.gz 67%[============> ] 190,84M 35,7MB/s eta 3s 1000G.tar.gz 69%[============> ] 196,58M 35,2MB/s eta 3s 1000G.tar.gz 72%[=============> ] 203,15M 35,0MB/s eta 2s 1000G.tar.gz 74%[=============> ] 210,30M 35,0MB/s eta 2s 1000G.tar.gz 77%[==============> ] 217,40M 34,9MB/s eta 2s 1000G.tar.gz 79%[==============> ] 224,59M 35,0MB/s eta 2s 1000G.tar.gz 82%[===============> ] 231,89M 35,0MB/s eta 2s 1000G.tar.gz 85%[================> ] 239,13M 35,0MB/s eta 1s 1000G.tar.gz 87%[================> ] 246,30M 35,0MB/s eta 1s 1000G.tar.gz 90%[=================> ] 253,51M 35,1MB/s eta 1s 1000G.tar.gz 92%[=================> ] 260,66M 35,2MB/s eta 1s 1000G.tar.gz 95%[==================> ] 267,75M 35,1MB/s eta 1s 1000G.tar.gz 97%[==================> ] 274,86M 35,1MB/s eta 0s 1000G.tar.gz 100%[===================>] 280,96M 35,1MB/s in 8,2s
2023-11-07 13:14:24 (34,4 MB/s) - ‘1000G.tar.gz’ saved [294605223/294605223]
For running the PCA and Admixture analyses with ANGSD, we will need to build a list of the available bam-files. It is also useful for us to check from this list what human populations are present in the Phase 1 of 1000G project.
In [3]:
cd 1000G_bam_files
find $PWD -name '*.bam' > ../1000G_bam_list.txt
cd ..
head 1000G_bam_list.txt/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA18543.mapped.ILLUMINA.bwa.CHB.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA20334.mapped.ILLUMINA.bwa.ASW.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA18633.mapped.ILLUMINA.bwa.CHB.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA12348.mapped.ILLUMINA.bwa.CEU.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA12842.mapped.ILLUMINA.bwa.CEU.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA11832.mapped.ILLUMINA.bwa.CEU.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA12275.mapped.ILLUMINA.bwa.CEU.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA19761.mapped.ILLUMINA.bwa.MXL.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA19213.mapped.ILLUMINA.bwa.YRI.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA18558.mapped.ILLUMINA.bwa.CHB.low_coverage.20130415.bam
In [5]:
wc -l 1000G_bam_list.txt435 1000G_bam_list.txt
We can see that in total our dataset includes 435 bam-files corresponding to African (YRI), European (CEU) and Asian (CHB) populations, as well as two admixed populations: African-American (ASW) and Mexican (MXL). ANGSD operates with genetic variation computed via genotype likelihoods. Therefore, we will have to perform genotype likelihoods variant calling prior to PCA and Admixture analyses. If you do not have ANGSD installed already, it can be downloaded and installed using instrictions from http://www.popgen.dk/angsd/index.php/Installation.
In [4]:
angsd -bam 1000G_bam_list.txt -GL 2 -doMajorMinor 1 -doMaf 1 -SNP_pval 2e-6 -minMapQ 30 -minQ 20 -minInd 25 -minMaf 0.05 -doGlf 2 -out 1000G -P 5 -> angsd version: 0.940-dirty (htslib: 1.16) build(Aug 23 2023 11:40:54)
-> angsd -bam 1000G_bam_list.txt -GL 2 -doMajorMinor 1 -doMaf 1 -SNP_pval 2e-6 -minMapQ 30 -minQ 20 -minInd 25 -minMaf 0.05 -doGlf 2 -out 1000G -P 5
-> Inputtype is BAM/CRAM
[multiReader] 435 samples in 435 input files
-> SNP-filter using a pvalue: 2.000000e-06 correspond to 22.595043 likelihood units
-> Parsing 435 number of samples
-> Allocated ~ 10 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 20 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 30 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 40 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 50 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 60 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 70 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 80 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 90 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 100 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 110 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 120 million nodes to the nodepool, this is not an estimate of the memory usage
-> Allocated ~ 130 million nodes to the nodepool, this is not an estimate of the memory usage
-> Printing at chr: 7 pos:20911210 chunknumber 100 contains 2 sites -> Printing at chr: 14 pos:20692360 chunknumber 200 contains 699 sites
-> Done reading data waiting for calculations to finish
-> Done waiting for threads
-> Output filenames:
->"1000G.arg"
->"1000G.beagle.gz"
->"1000G.mafs.gz"
-> Tue Nov 7 13:38:46 2023
-> Arguments and parameters for all analysis are located in .arg file
-> Total number of sites analyzed: 167518
-> Number of sites retained after filtering: 1306
[ALL done] cpu-time used = 150.90 sec
[ALL done] walltime used = 61.00 sec
The calculated genotype likelihoods for 106 variable sites are contained within the 1000G.beagle.gz file, which we are now going to use for performing PCA analysis. The second returned file, 1000G.mafs.gz, contains information about allele frequencies of the identified genetic variants. You can read about advantages of doing PCA via genotype likelihoods for example here. The PCANGSD method can be installed separately from ANGSD from https://github.com/Rosemeis/pcangsd.
In [5]:
pcangsd -b 1000G.beagle.gz -o pca_1000G -t 4-------------------------------------
PCAngsd v1.2
Jonas Meisner and Anders Albrechtsen.
Using 4 thread(s).
-------------------------------------
Parsing Beagle file.
Loaded 1306 sites and 435 individuals.
Estimating minor allele frequencies.
EM (MAF) converged at iteration: 16
Number of sites after MAF filtering (0.05): 1306
Estimating covariance matrix.
Using 15 principal components (MAP test).
Individual allele frequencies estimated (1).
Individual allele frequencies estimated (2). RMSE=0.066385679
Individual allele frequencies estimated (3). RMSE=0.050454147
Individual allele frequencies estimated (4). RMSE=0.006292501
Individual allele frequencies estimated (5). RMSE=0.025063241
Individual allele frequencies estimated (6). RMSE=0.003102337
Individual allele frequencies estimated (7). RMSE=0.024891028
Individual allele frequencies estimated (8). RMSE=0.002931002
Individual allele frequencies estimated (9). RMSE=0.024905935
Individual allele frequencies estimated (10). RMSE=0.003440379
Individual allele frequencies estimated (11). RMSE=0.024913993
Individual allele frequencies estimated (12). RMSE=0.002420334
Individual allele frequencies estimated (13). RMSE=0.002137294
Individual allele frequencies estimated (14). RMSE=0.001955432
Individual allele frequencies estimated (15). RMSE=0.001708493
Individual allele frequencies estimated (16). RMSE=0.001492948
Individual allele frequencies estimated (17). RMSE=0.001377949
Individual allele frequencies estimated (18). RMSE=0.024781208
Individual allele frequencies estimated (19). RMSE=0.001125538
Individual allele frequencies estimated (20). RMSE=0.02478423
Individual allele frequencies estimated (21). RMSE=0.000906437
Individual allele frequencies estimated (22). RMSE=0.000914994
Individual allele frequencies estimated (23). RMSE=0.000651902
Individual allele frequencies estimated (24). RMSE=0.000590639
Individual allele frequencies estimated (25). RMSE=0.000566323
Individual allele frequencies estimated (26). RMSE=0.024761193
Individual allele frequencies estimated (27). RMSE=0.000427994
Individual allele frequencies estimated (28). RMSE=0.000424277
Individual allele frequencies estimated (29). RMSE=0.00039096
Individual allele frequencies estimated (30). RMSE=0.000348833
Individual allele frequencies estimated (31). RMSE=0.000310296
Individual allele frequencies estimated (32). RMSE=0.000292033
Individual allele frequencies estimated (33). RMSE=0.000268337
Individual allele frequencies estimated (34). RMSE=0.024759974
Individual allele frequencies estimated (35). RMSE=0.000225774
Individual allele frequencies estimated (36). RMSE=0.000209186
Individual allele frequencies estimated (37). RMSE=0.000194486
Individual allele frequencies estimated (38). RMSE=0.000179259
Individual allele frequencies estimated (39). RMSE=0.000164353
Individual allele frequencies estimated (40). RMSE=0.000144426
Individual allele frequencies estimated (41). RMSE=0.000147983
Individual allele frequencies estimated (42). RMSE=0.000143973
Individual allele frequencies estimated (43). RMSE=0.000138792
Individual allele frequencies estimated (44). RMSE=0.000135654
Individual allele frequencies estimated (45). RMSE=0.000153877
Individual allele frequencies estimated (46). RMSE=0.000120664
Individual allele frequencies estimated (47). RMSE=0.000112655
Individual allele frequencies estimated (48). RMSE=0.000110528
Individual allele frequencies estimated (49). RMSE=0.000133146
Individual allele frequencies estimated (50). RMSE=0.000102072
Individual allele frequencies estimated (51). RMSE=0.000100332
Individual allele frequencies estimated (52). RMSE=0.024759417
Individual allele frequencies estimated (53). RMSE=8.5162e-05
Individual allele frequencies estimated (54). RMSE=8.0157e-05
Individual allele frequencies estimated (55). RMSE=7.5776e-05
Individual allele frequencies estimated (56). RMSE=7.2006e-05
Individual allele frequencies estimated (57). RMSE=0.024759391
Individual allele frequencies estimated (58). RMSE=6.5975e-05
Individual allele frequencies estimated (59). RMSE=6.2556e-05
Individual allele frequencies estimated (60). RMSE=6.0224e-05
Individual allele frequencies estimated (61). RMSE=5.7719e-05
Individual allele frequencies estimated (62). RMSE=5.5379e-05
Individual allele frequencies estimated (63). RMSE=5.3232e-05
Individual allele frequencies estimated (64). RMSE=5.0864e-05
Individual allele frequencies estimated (65). RMSE=4.9148e-05
Individual allele frequencies estimated (66). RMSE=4.7062e-05
Individual allele frequencies estimated (67). RMSE=4.5683e-05
Individual allele frequencies estimated (68). RMSE=4.4471e-05
Individual allele frequencies estimated (69). RMSE=5.0811e-05
Individual allele frequencies estimated (70). RMSE=4.275e-05
Individual allele frequencies estimated (71). RMSE=4.1382e-05
Individual allele frequencies estimated (72). RMSE=4.005e-05
Individual allele frequencies estimated (73). RMSE=3.9036e-05
Individual allele frequencies estimated (74). RMSE=3.7008e-05
Individual allele frequencies estimated (75). RMSE=3.5606e-05
Individual allele frequencies estimated (76). RMSE=3.4998e-05
Individual allele frequencies estimated (77). RMSE=3.3238e-05
Individual allele frequencies estimated (78). RMSE=3.3203e-05
Individual allele frequencies estimated (79). RMSE=3.0969e-05
Individual allele frequencies estimated (80). RMSE=3.0605e-05
Individual allele frequencies estimated (81). RMSE=2.9137e-05
Individual allele frequencies estimated (82). RMSE=2.8265e-05
Individual allele frequencies estimated (83). RMSE=2.8226e-05
Individual allele frequencies estimated (84). RMSE=2.6588e-05
Individual allele frequencies estimated (85). RMSE=2.6364e-05
Individual allele frequencies estimated (86). RMSE=2.5135e-05
Individual allele frequencies estimated (87). RMSE=2.4293e-05
Individual allele frequencies estimated (88). RMSE=2.3529e-05
Individual allele frequencies estimated (89). RMSE=2.2974e-05
Individual allele frequencies estimated (90). RMSE=2.1807e-05
Individual allele frequencies estimated (91). RMSE=2.1036e-05
Individual allele frequencies estimated (92). RMSE=2.1025e-05
Individual allele frequencies estimated (93). RMSE=2.0272e-05
Individual allele frequencies estimated (94). RMSE=1.9758e-05
Individual allele frequencies estimated (95). RMSE=1.9355e-05
Individual allele frequencies estimated (96). RMSE=1.8164e-05
Individual allele frequencies estimated (97). RMSE=1.8058e-05
Individual allele frequencies estimated (98). RMSE=1.7484e-05
Individual allele frequencies estimated (99). RMSE=1.7081e-05
Individual allele frequencies estimated (100). RMSE=1.6462e-05
Individual allele frequencies estimated (101). RMSE=1.6259e-05
PCAngsd did not converge!
Saved covariance matrix as pca_1000G.cov
Total elapsed time: 0m3s
A peculiarity of computing PCA via ANGSD is that it does not directly generates a PCA plot but returns a variance-covariance matrix pca_1000G.cov which you will have to manually eigen-value-decompose and plot the eigen vectors in R. Note, that in order to color data points on the PCA plot, we will parse the list of bam-files from above in R, and extract the populations labels.
In [9]:
setwd("/home/nikolay/Documents/Teaching/PopGen_2023/population_structure")
C <- as.matrix(read.table("pca_1000G.cov"))
e <- eigen(C)
pops <- readLines("1000G_bam_list.txt")
pops <- sapply(strsplit(pops, "\\."), function(x) x[6])
mycolor <- rep("red", length(pops))
mycolor[pops == "CEU"] <- "blue"
mycolor[pops == "CHB"] <- "green"
mycolor[pops == "MXL"] <- "brown"
mycolor[pops == "ASW"] <- "magenta"
plot(e$vectors[, 1:2], xlab = "PC1", ylab = "PC2", main = "PCA 1000G Project", col = mycolor, pch = 19)
legend("bottomright", c("YRI", "CEU", "CHB", "MXL", "ASW"), fill = c("red", "blue", "green", "brown", "magenta"), cex = 1, inset = 0.02)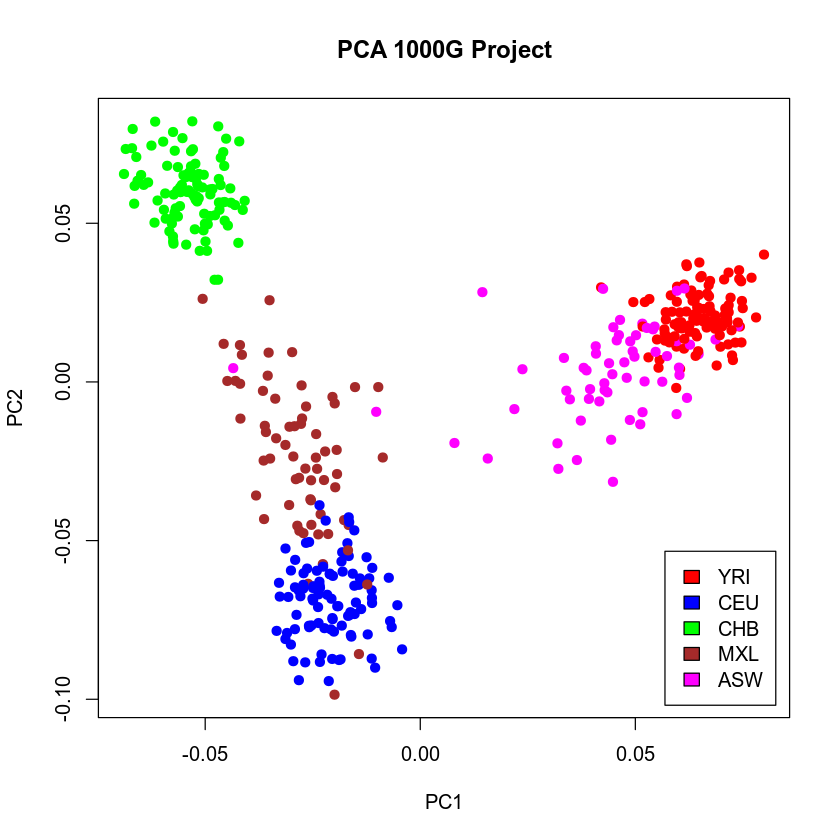
As expected, we have three clear clusters (main sources of variation) corresponding to African (YRI), European (CEU) and Asian (CHB) populations, with the admixed populations falling in between. Let us see that this conclusion can be further confirmed from the Admixture analysis in the next section.
In this section, we will perform admixture analysis using the genetic variation data from 1000G project identified in the previous section, i.e. the file 1000G.beagle.gz. We will keep using the genotype likelihoods approach with ANGSD, and utilize NGSadmix, which is a tool within ANGSD. You do not need to install NGSadmix separetely as it is installed together with ANGSD, however, in case there are problems, the NGSadmix executable can be found within pcangsd/misc and launched from there. Please note that below we explicitly ask NGSadmix to detect K = 3 clusters.
In [12]:
NGSadmix -likes 1000G.beagle.gz -K 3 -minMaf 0.05 -seed 1 -o 1000GInput: lname=1000G.beagle.gz nPop=3, fname=(null) qname=(null) outfiles=1000G
Setup: seed=1 nThreads=1 method=1
Convergence: maxIter=2000 tol=0.000010 tolLike50=0.100000 dymBound=0
Filters: misTol=0.050000 minMaf=0.050000 minLrt=0.000000 minInd=0
Input file has dim: nsites=1306 nind=435
Input file has dim (AFTER filtering): nsites=1306 nind=435
iter[start] like is=725129.713622
iter[50] like is=-454493.026878 thres=0.000841
iter[100] like is=-454474.788034 thres=0.000032
EM accelerated has reached convergence with tol 0.000010
best like=-454474.752968 after 115 iterations
-> Dumpedfiles are: 1000G.log
-> Dumpedfiles are: 1000G.qopt
-> Dumpedfiles are: 1000G.fopt.gz
[ALL done] cpu-time used = 4.84 sec
[ALL done] walltime used = 5.00 sec
NGSadmix produces two files: 1000G.qopt and 1000G.fopt.gz containing admixture proportions and allele frequencies, respectively. If we look inside the 1000G.qopt files, we will see that for eahc of the 435 data points it contains fractions (i.e. contributions) of each of the 3 clusters that we required NGSadmix to identify.
In [6]:
head 1000G.qopt0.00543120337380969750 0.03047764250291678037 0.96409115412327339723
0.39746657097866411323 0.59699325992555196674 0.00554016909578402498
0.00291473396727394071 0.00000000099999999999 0.99708526503272598696
0.89272766803245084954 0.00000008404621942802 0.10727224792132973585
0.87148184717709176184 0.12851815182290812767 0.00000000099999999997
0.81529873054609225402 0.15780315987658605215 0.02689810957732174240
0.83248589650022153386 0.07729402969534844570 0.09022007380443013147
0.71004516603356415683 0.00000000099999999998 0.28995483296643576043
0.09294217159488990521 0.90705782740511009532 0.00000000099999999997
0.00000000099999999994 0.00000000099999999994 0.99999999799999994554
Finally, we will plot the results using some helping functions from the ANGSD and NGSadmix developers.
In [26]:
setwd("/home/nikolay/Documents/Teaching/PopGen_2023/population_structure")
pops <- readLines("1000G_bam_list.txt")
pops <- sapply(strsplit(pops, "\\."), function(x) x[6])
source("https://raw.githubusercontent.com/GenisGE/evalAdmix/master/visFuns.R")
qopts <- read.table("1000G.qopt")
ord <- orderInds(pop = pops, q = qopts, popord = c("YRI", "ASW", "CEU", "MXL", "CHB"))
barplot(t(qopts)[, ord], col = c(3, 2, 4), las = 2, space = 0, border = NA)
text(sort(tapply(1:length(pops), pops[ord], mean)), 0.1, unique(pops[ord]))
abline(v = cumsum(sapply(unique(pops[ord]), function(x) {sum(pops[ord] == x)})), col = 1, lwd = 1.2)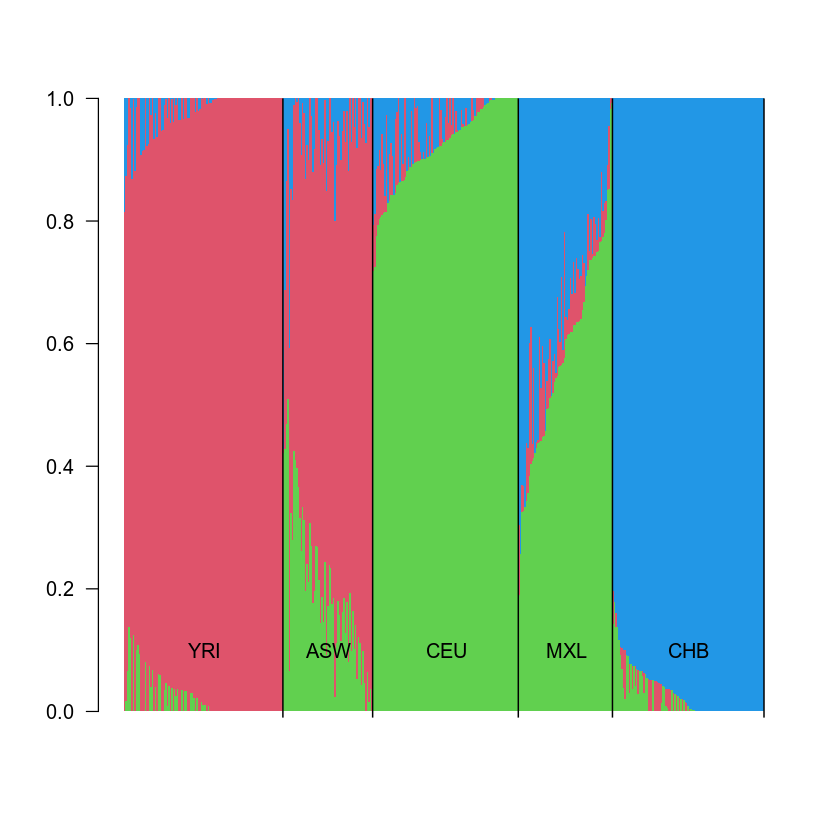
Again, we see three nearly unadmixed populations: YRI, CHB and CEU, and two admixed populations: MXL (mixture of CEU and CHB) and ASW (mixture of YRI and CEU). Therefore, one can infer for each individual, how much this individual is admixed and what are the populations contributing to the admixture.
In this section, we will perform Fst analysis via genotype likelihoods using the 1000G data, i.e. we will compare allele frequencies between European (CEU) and African (YRI) populations. The way ANGSD does it, it first computes Site Frequency Spectra (SFS) for CEU and YRI individually. So we will have to subset the list of bam-files to CEU and YRI individuals only and compute SFS. Below, we will start with CEU:
In [11]:
grep CEU 1000G_bam_list.txt > 1000G_CEU_bam_list.txt
wc -l 1000G_CEU_bam_list.txt
head 1000G_CEU_bam_list.txt99 1000G_CEU_bam_list.txt
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA12348.mapped.ILLUMINA.bwa.CEU.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA12842.mapped.ILLUMINA.bwa.CEU.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA11832.mapped.ILLUMINA.bwa.CEU.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA12275.mapped.ILLUMINA.bwa.CEU.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA11830.mapped.ILLUMINA.bwa.CEU.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA12777.mapped.ILLUMINA.bwa.CEU.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA12287.mapped.ILLUMINA.bwa.CEU.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA07357.mapped.ILLUMINA.bwa.CEU.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA12286.mapped.ILLUMINA.bwa.CEU.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA11930.mapped.ILLUMINA.bwa.CEU.low_coverage.20130415.bam
In [15]:
angsd -b 1000G_CEU_bam_list.txt -anc anc.fa -out CEU -dosaf 1 -gl 1 -> angsd version: 0.940-dirty (htslib: 1.16) build(Aug 23 2023 11:40:54)
-> angsd -b 1000G_CEU_bam_list.txt -anc anc.fa -out CEU -dosaf 1 -gl 1
-> Inputtype is BAM/CRAM
[multiReader] 99 samples in 99 input files
-> Reading fasta: anc.fa
-> Parsing 99 number of samples
-> Printing at chr: 7 pos:20690960 chunknumber 100 contains 621 sites -> Printing at chr: 13 pos:20676861 chunknumber 200 contains 833 sites
-> Done reading data waiting for calculations to finish
-> Done waiting for threads
-> Output filenames:
->"CEU.arg"
->"CEU.saf.gz"
->"CEU.saf.pos.gz"
->"CEU.saf.idx"
-> Tue Nov 7 14:21:40 2023
-> Arguments and parameters for all analysis are located in .arg file
-> Total number of sites analyzed: 163347
-> Number of sites retained after filtering: 162624
[ALL done] cpu-time used = 41.99 sec
[ALL done] walltime used = 42.00 sec
Now, we will subset YRI individuals and compute SFS for the African population.
In [12]:
grep YRI 1000G_bam_list.txt > 1000G_YRI_bam_list.txt
wc -l 1000G_YRI_bam_list.txt
head 1000G_YRI_bam_list.txt108 1000G_YRI_bam_list.txt
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA19213.mapped.ILLUMINA.bwa.YRI.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA18504.mapped.ILLUMINA.bwa.YRI.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA19108.mapped.ILLUMINA.bwa.YRI.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA19238.mapped.ILLUMINA.bwa.YRI.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA19204.mapped.ILLUMINA.bwa.YRI.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA18516.mapped.ILLUMINA.bwa.YRI.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA18878.mapped.ILLUMINA.bwa.YRI.low_coverage.20130415.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA18874.mapped.ILLUMINA.bwa.YRI.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA18488.mapped.ILLUMINA.bwa.YRI.low_coverage.20120522.bam
/home/nikolay/Documents/Teaching/PopGen_2023/population_structure/1000G_bam_files/small.NA18861.mapped.ILLUMINA.bwa.YRI.low_coverage.20130415.bam
In [16]:
angsd -b 1000G_YRI_bam_list.txt -anc anc.fa -out YRI -dosaf 1 -gl 1 -> angsd version: 0.940-dirty (htslib: 1.16) build(Aug 23 2023 11:40:54)
-> angsd -b 1000G_YRI_bam_list.txt -anc anc.fa -out YRI -dosaf 1 -gl 1
-> Inputtype is BAM/CRAM
[multiReader] 108 samples in 108 input files
-> Reading fasta: anc.fa
-> Parsing 108 number of samples
-> Printing at chr: 8 pos:20152114 chunknumber 100 contains 597 sites -> Printing at chr: 16 pos:20080879 chunknumber 200 contains 601 sites
-> Done reading data waiting for calculations to finish
-> Done waiting for threads
-> Output filenames:
->"YRI.arg"
->"YRI.saf.gz"
->"YRI.saf.pos.gz"
->"YRI.saf.idx"
-> Tue Nov 7 14:22:50 2023
-> Arguments and parameters for all analysis are located in .arg file
-> Total number of sites analyzed: 159100
-> Number of sites retained after filtering: 158453
[ALL done] cpu-time used = 44.02 sec
[ALL done] walltime used = 44.00 sec
In [17]:
realSFS CEU.saf.idx YRI.saf.idx > CEU.YRI.ml[persaf::persaf_init] Version of CEU.saf.idx is 3
[persaf::persaf_init] Assuming .saf.gz file is CEU.saf.gz
[persaf::persaf_init] Assuming .saf.pos.gz file is CEU.saf.pos.gz
[persaf::persaf_init] Version of YRI.saf.idx is 3
[persaf::persaf_init] Assuming .saf.gz file is YRI.saf.gz
[persaf::persaf_init] Assuming .saf.pos.gz file is YRI.saf.pos.gz
-> args: tole:0.000000 nthreads:4 maxiter:100 nsites(block):0 start:(null) chr:(null) start:-1 stop:-1 fstout:(null) oldout:0 seed:-1 bootstrap:0 resample_chr:0 whichFst:0 fold:0 ref:(null) anc:(null)
[main] Multi SFS is 'still' under development. Please report strange behaviour.
-> The choice of -nSites will require atleast: 45.203266 megabyte memory, that is at least: 0.28% of total memory
-> dim(CEU.saf.idx):199
-> dim(YRI.saf.idx):217
-> Dimension of parameter space: 43183
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:1
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[1] from pop0: 11218
-> Sites to keep[1] from pop1: 11218
-> [readdata] lastread:11218 posi:20017990
-> Comparing positions: 1 with 0 has:11218
-> Only read nSites: 11218 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:10
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[10] from pop0: 10005
-> Sites to keep[10] from pop1: 10005
-> [readdata] lastread:10005 posi:20017990
-> Comparing positions: 1 with 0 has:21223
-> Only read nSites: 21223 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:11
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[11] from pop0: 11825
-> Sites to keep[11] from pop1: 11825
-> [readdata] lastread:11825 posi:20017990
-> Comparing positions: 1 with 0 has:33048
-> Only read nSites: 33048 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:12
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[12] from pop0: 7433
-> Sites to keep[12] from pop1: 7433
-> [readdata] lastread:7433 posi:20017990
-> Comparing positions: 1 with 0 has:40481
-> Only read nSites: 40481 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:13
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[13] from pop0: 4580
-> Sites to keep[13] from pop1: 4580
-> [readdata] lastread:4580 posi:20017990
-> Comparing positions: 1 with 0 has:45061
-> Only read nSites: 45061 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:14
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[14] from pop0: 4269
-> Sites to keep[14] from pop1: 4269
-> [readdata] lastread:4269 posi:20017990
-> Comparing positions: 1 with 0 has:49330
-> Only read nSites: 49330 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:16
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[16] from pop0: 8855
-> Sites to keep[16] from pop1: 8855
-> [readdata] lastread:8855 posi:20017990
-> Comparing positions: 1 with 0 has:58185
-> Only read nSites: 58185 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:17
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[17] from pop0: 1564
-> Sites to keep[17] from pop1: 1564
-> [readdata] lastread:1564 posi:20017990
-> Comparing positions: 1 with 0 has:59749
-> Only read nSites: 59749 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:18
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[18] from pop0: 6271
-> Sites to keep[18] from pop1: 6271
-> [readdata] lastread:6271 posi:20017990
-> Comparing positions: 1 with 0 has:66020
-> Only read nSites: 66020 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:19
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[19] from pop0: 1406
-> Sites to keep[19] from pop1: 1406
-> [readdata] lastread:1406 posi:20017990
-> Comparing positions: 1 with 0 has:67426
-> Only read nSites: 67426 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:2
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[2] from pop0: 9853
-> Sites to keep[2] from pop1: 9853
-> [readdata] lastread:9853 posi:20017990
-> Comparing positions: 1 with 0 has:77279
-> Only read nSites: 77279 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:20
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[20] from pop0: 11636
-> Sites to keep[20] from pop1: 11636
-> [readdata] lastread:11636 posi:20017990
-> Comparing positions: 1 with 0 has:88915
-> Only read nSites: 88915 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:21
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[21] from pop0: 7716
-> Sites to keep[21] from pop1: 7716
-> [readdata] lastread:7716 posi:20017990
-> Comparing positions: 1 with 0 has:96631
-> Only read nSites: 96631 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:22
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[22] from pop0: 3014
-> Sites to keep[22] from pop1: 3014
-> [readdata] lastread:3014 posi:20017990
-> Comparing positions: 1 with 0 has:99645
-> Only read nSites: 99645 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:3
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[3] from pop0: 7606
-> Sites to keep[3] from pop1: 7606
-> [readdata] lastread:7606 posi:20017990
-> Comparing positions: 1 with 0 has:107251
-> Only read nSites: 107251 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:4
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[4] from pop0: 6424
-> Sites to keep[4] from pop1: 6424
-> [readdata] lastread:6424 posi:20017990
-> Comparing positions: 1 with 0 has:113675
-> Only read nSites: 113675 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:5
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[5] from pop0: 3210
-> Sites to keep[5] from pop1: 3210
-> [readdata] lastread:3210 posi:20017990
-> Comparing positions: 1 with 0 has:116885
-> Only read nSites: 116885 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:6
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[6] from pop0: 9615
-> Sites to keep[6] from pop1: 9615
-> [readdata] lastread:9615 posi:20017990
-> Comparing positions: 1 with 0 has:126500
-> Only read nSites: 126500 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:7
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[7] from pop0: 8824
-> Sites to keep[7] from pop1: 8824
-> [readdata] lastread:8824 posi:20017990
-> Comparing positions: 1 with 0 has:135324
-> Only read nSites: 135324 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:8
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[8] from pop0: 16633
-> Sites to keep[8] from pop1: 16633
-> [readdata] lastread:16633 posi:20017990
-> Comparing positions: 1 with 0 has:151957
-> Only read nSites: 151957 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Is in multi sfs, will now read data from chr:9
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[9] from pop0: 6247
-> Sites to keep[9] from pop1: 6247
-> [readdata] lastread:6247 posi:20017990
-> Comparing positions: 1 with 0 has:158204
-> Only read nSites: 158204 will therefore prepare next chromosome (or exit)
-> Done reading data from chromosome will prepare next chromosome
-> Will run optimization on nSites: 158204
------------
startlik=-1500482.960971
lik[2]=-51446.465409 diff=1.449036e+06 alpha:1.000000 sr2:2.565215e-01 nsites_difference[0]: 1.304900e+05
-> Instability detected, accelerated guess is too close to bound or outside will fallback to regular EM for this step
lik[4]=-36952.653747 diff=1.449381e+04 alpha:1.520022 sr2:1.189258e-02 nsites_difference[0]: 2.079652e+04
lik[6]=-35693.960846 diff=1.258693e+03 alpha:1.000000 sr2:1.954655e-04 nsites_difference[0]: 2.559705e+03
lik[8]=-35550.497375 diff=1.434635e+02 alpha:1.802902 sr2:5.252347e-06 nsites_difference[0]: 4.155786e+02
lik[10]=-35515.651398 diff=3.484598e+01 alpha:1.000000 sr2:2.383104e-07 nsites_difference[0]: 8.639444e+01
lik[12]=-35497.489801 diff=1.816160e+01 alpha:2.286558 sr2:1.786152e-08 nsites_difference[1]: 2.290752e+01
lik[14]=-35485.181049 diff=1.230875e+01 alpha:1.000000 sr2:2.166627e-09 nsites_difference[1]: 8.282941e+00
lik[16]=-35476.102765 diff=9.078283e+00 alpha:3.702718 sr2:4.923696e-10 nsites_difference[1]: 3.370521e+00
lik[18]=-35469.117497 diff=6.985269e+00 alpha:1.000000 sr2:2.181576e-10 nsites_difference[7]: 1.982587e+00
lik[20]=-35463.588847 diff=5.528650e+00 alpha:4.000000 sr2:1.404275e-10 nsites_difference[7]: 1.666359e+00
lik[22]=-35459.119268 diff=4.469579e+00 alpha:1.000000 sr2:1.036972e-10 nsites_difference[7]: 1.395785e+00
lik[24]=-35455.444426 diff=3.674842e+00 alpha:4.000000 sr2:8.067148e-11 nsites_difference[7]: 1.171575e+00
lik[26]=-35452.380826 diff=3.063601e+00 alpha:1.000000 sr2:6.443842e-11 nsites_difference[7]: 9.876003e-01
lik[28]=-35449.796828 diff=2.583998e+00 alpha:4.000000 sr2:5.239715e-11 nsites_difference[7]: 8.364649e-01
lik[30]=-35447.595469 diff=2.201359e+00 alpha:1.000000 sr2:4.321692e-11 nsites_difference[7]: 7.116037e-01
lik[32]=-35445.703737 diff=1.891731e+00 alpha:4.000000 sr2:3.608661e-11 nsites_difference[11]: 6.311199e-01
lik[34]=-35444.065627 diff=1.638110e+00 alpha:1.000000 sr2:3.046627e-11 nsites_difference[11]: 5.900838e-01
lik[36]=-35442.637478 diff=1.428149e+00 alpha:4.000000 sr2:2.597932e-11 nsites_difference[11]: 5.522152e-01
lik[38]=-35441.384775 diff=1.252703e+00 alpha:1.000000 sr2:2.235567e-11 nsites_difference[11]: 5.171917e-01
lik[40]=-35440.279895 diff=1.104880e+00 alpha:4.000000 sr2:1.939778e-11 nsites_difference[11]: 4.847370e-01
lik[42]=-35439.300487 diff=9.794077e-01 alpha:1.000000 sr2:1.695900e-11 nsites_difference[11]: 4.546117e-01
lik[44]=-35438.428292 diff=8.721947e-01 alpha:4.000000 sr2:1.492908e-11 nsites_difference[11]: 4.266064e-01
lik[46]=-35437.648264 diff=7.800289e-01 alpha:1.000000 sr2:1.322427e-11 nsites_difference[11]: 4.005367e-01
lik[48]=-35436.947904 diff=7.003600e-01 alpha:4.000000 sr2:1.178030e-11 nsites_difference[11]: 3.762390e-01
lik[50]=-35436.316761 diff=6.311430e-01 alpha:1.000000 sr2:1.054743e-11 nsites_difference[11]: 3.535673e-01
lik[52]=-35435.746037 diff=5.707233e-01 alpha:4.000000 sr2:9.486871e-12 nsites_difference[16]: 3.430160e-01
lik[54]=-35435.228286 diff=5.177516e-01 alpha:1.000000 sr2:8.568103e-12 nsites_difference[16]: 3.329148e-01
lik[56]=-35434.757167 diff=4.711190e-01 alpha:4.000000 sr2:7.766956e-12 nsites_difference[16]: 3.230556e-01
lik[58]=-35434.327258 diff=4.299086e-01 alpha:1.000000 sr2:7.064145e-12 nsites_difference[16]: 3.134433e-01
lik[60]=-35433.933901 diff=3.933572e-01 alpha:4.000000 sr2:6.444171e-12 nsites_difference[16]: 3.040804e-01
lik[62]=-35433.573075 diff=3.608262e-01 alpha:1.000000 sr2:5.894488e-12 nsites_difference[16]: 2.949677e-01
lik[64]=-35433.241296 diff=3.317786e-01 alpha:4.000000 sr2:5.404870e-12 nsites_difference[16]: 2.861046e-01
lik[66]=-35432.935536 diff=3.057603e-01 alpha:1.000000 sr2:4.966920e-12 nsites_difference[16]: 2.774893e-01
lik[68]=-35432.653149 diff=2.823863e-01 alpha:4.000000 sr2:4.573696e-12 nsites_difference[16]: 2.691193e-01
lik[70]=-35432.391821 diff=2.613280e-01 alpha:1.000000 sr2:4.219417e-12 nsites_difference[16]: 2.609914e-01
lik[72]=-35432.149517 diff=2.423047e-01 alpha:4.000000 sr2:3.899235e-12 nsites_difference[16]: 2.531018e-01
lik[74]=-35431.924441 diff=2.250752e-01 alpha:1.000000 sr2:3.609056e-12 nsites_difference[16]: 2.454464e-01
lik[76]=-35431.715010 diff=2.094317e-01 alpha:4.000000 sr2:3.345401e-12 nsites_difference[16]: 2.380208e-01
lik[78]=-35431.519815 diff=1.951947e-01 alpha:1.000000 sr2:3.105296e-12 nsites_difference[16]: 2.308202e-01
lik[80]=-35431.337606 diff=1.822086e-01 alpha:4.000000 sr2:2.886179e-12 nsites_difference[16]: 2.238396e-01
lik[82]=-35431.167268 diff=1.703380e-01 alpha:1.000000 sr2:2.685833e-12 nsites_difference[16]: 2.170740e-01
lik[84]=-35431.007803 diff=1.594649e-01 alpha:4.000000 sr2:2.502330e-12 nsites_difference[16]: 2.105180e-01
lik[86]=-35430.858317 diff=1.494862e-01 alpha:1.000000 sr2:2.333982e-12 nsites_difference[16]: 2.041665e-01
lik[88]=-35430.718006 diff=1.403111e-01 alpha:4.000000 sr2:2.179306e-12 nsites_difference[16]: 1.980140e-01
lik[90]=-35430.586146 diff=1.318602e-01 alpha:1.000000 sr2:2.036993e-12 nsites_difference[16]: 1.920553e-01
lik[92]=-35430.462083 diff=1.240630e-01 alpha:4.000000 sr2:1.905885e-12 nsites_difference[16]: 1.862849e-01
lik[94]=-35430.345226 diff=1.168575e-01 alpha:1.000000 sr2:1.784948e-12 nsites_difference[16]: 1.806975e-01
lik[96]=-35430.235037 diff=1.101884e-01 alpha:4.000000 sr2:1.673264e-12 nsites_difference[16]: 1.752880e-01
lik[98]=-35430.131030 diff=1.040069e-01 alpha:1.000000 sr2:1.570008e-12 nsites_difference[16]: 1.700509e-01
lik[100]=-35430.032761 diff=9.826906e-02 alpha:4.000000 sr2:1.474443e-12 nsites_difference[16]: 1.649813e-01
likelihood: -35430.032761
------------
-> Done reading data from chromosome will prepare next chromosome
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> NB output is no longer log probs of the frequency spectrum!
-> Output is now simply the expected values!
-> You can convert to the old format simply with log(norm(x))
-> Please check that it has converged!
-> You can add start new optimization by supplying -sfs FILE, where is >FILE from this run
-> -maxIter INT -tole FLOAT
In [22]:
realSFS fst index CEU.saf.idx YRI.saf.idx -sfs CEU.YRI.ml -fstout CEU_YRI[persaf::persaf_init] Version of CEU.saf.idx is 3
[persaf::persaf_init] Assuming .saf.gz file is CEU.saf.gz
[persaf::persaf_init] Assuming .saf.pos.gz file is CEU.saf.pos.gz
[persaf::persaf_init] Version of YRI.saf.idx is 3
[persaf::persaf_init] Assuming .saf.gz file is YRI.saf.gz
[persaf::persaf_init] Assuming .saf.pos.gz file is YRI.saf.pos.gz
-> args: tole:0.000000 nthreads:4 maxiter:100 nsites(block):0 start:CEU.YRI.ml chr:(null) start:-1 stop:-1 fstout:CEU_YRI oldout:0 seed:-1 bootstrap:0 resample_chr:0 whichFst:0 fold:0 ref:(null) anc:(null)
-> nSites: 100000
-> IMPORTANT: please make sure that your saf files haven't been folded with -fold 1 in -doSaf in angsd
-> [reynoldFst] sfs1:198 sfs2:216 dimspace:43183
-> generating offset remapper lookup
-> isSame:1 adjusting foldfactors
-> Reading: CEU.YRI.ml assuming counts (will normalize to probs internally)
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[1] from pop0: 11218
-> Sites to keep[1] from pop1: 11218
-> [readdata] lastread:11218 posi:20017990
-> Comparing positions: 1 with 0 has:11218
-> Will now do fst temp dump using a chunk of 11218
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[10] from pop0: 10005
-> Sites to keep[10] from pop1: 10005
-> [readdata] lastread:10005 posi:20004692
-> Comparing positions: 1 with 0 has:10005
-> Will now do fst temp dump using a chunk of 10005
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[11] from pop0: 11825
-> Sites to keep[11] from pop1: 11825
-> [readdata] lastread:11825 posi:20009117
-> Comparing positions: 1 with 0 has:11825
-> Will now do fst temp dump using a chunk of 11825
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[12] from pop0: 7433
-> Sites to keep[12] from pop1: 7433
-> [readdata] lastread:7433 posi:20008763
-> Comparing positions: 1 with 0 has:7433
-> Will now do fst temp dump using a chunk of 7433
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[13] from pop0: 4580
-> Sites to keep[13] from pop1: 4580
-> [readdata] lastread:4580 posi:20147408
-> Comparing positions: 1 with 0 has:4580
-> Will now do fst temp dump using a chunk of 4580
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[14] from pop0: 4269
-> Sites to keep[14] from pop1: 4269
-> [readdata] lastread:4269 posi:20435704
-> Comparing positions: 1 with 0 has:4269
-> Will now do fst temp dump using a chunk of 4269
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[16] from pop0: 8855
-> Sites to keep[16] from pop1: 8855
-> [readdata] lastread:8855 posi:20019440
-> Comparing positions: 1 with 0 has:8855
-> Will now do fst temp dump using a chunk of 8855
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[17] from pop0: 1564
-> Sites to keep[17] from pop1: 1564
-> [readdata] lastread:1564 posi:20012246
-> Comparing positions: 1 with 0 has:1564
-> Will now do fst temp dump using a chunk of 1564
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[18] from pop0: 6271
-> Sites to keep[18] from pop1: 6271
-> [readdata] lastread:6271 posi:20015325
-> Comparing positions: 1 with 0 has:6271
-> Will now do fst temp dump using a chunk of 6271
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[19] from pop0: 1406
-> Sites to keep[19] from pop1: 1406
-> [readdata] lastread:1406 posi:20061451
-> Comparing positions: 1 with 0 has:1406
-> Will now do fst temp dump using a chunk of 1406
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[2] from pop0: 9853
-> Sites to keep[2] from pop1: 9853
-> [readdata] lastread:9853 posi:20016996
-> Comparing positions: 1 with 0 has:9853
-> Will now do fst temp dump using a chunk of 9853
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[20] from pop0: 11636
-> Sites to keep[20] from pop1: 11636
-> [readdata] lastread:11636 posi:20014854
-> Comparing positions: 1 with 0 has:11636
-> Will now do fst temp dump using a chunk of 11636
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[21] from pop0: 7716
-> Sites to keep[21] from pop1: 7716
-> [readdata] lastread:7716 posi:20023741
-> Comparing positions: 1 with 0 has:7716
-> Will now do fst temp dump using a chunk of 7716
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[22] from pop0: 3014
-> Sites to keep[22] from pop1: 3014
-> [readdata] lastread:3014 posi:20010629
-> Comparing positions: 1 with 0 has:3014
-> Will now do fst temp dump using a chunk of 3014
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[3] from pop0: 7606
-> Sites to keep[3] from pop1: 7606
-> [readdata] lastread:7606 posi:20003128
-> Comparing positions: 1 with 0 has:7606
-> Will now do fst temp dump using a chunk of 7606
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[4] from pop0: 6424
-> Sites to keep[4] from pop1: 6424
-> [readdata] lastread:6424 posi:20064982
-> Comparing positions: 1 with 0 has:6424
-> Will now do fst temp dump using a chunk of 6424
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[5] from pop0: 3210
-> Sites to keep[5] from pop1: 3210
-> [readdata] lastread:3210 posi:20115852
-> Comparing positions: 1 with 0 has:3210
-> Will now do fst temp dump using a chunk of 3210
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[6] from pop0: 9615
-> Sites to keep[6] from pop1: 9615
-> [readdata] lastread:9615 posi:20001826
-> Comparing positions: 1 with 0 has:9615
-> Will now do fst temp dump using a chunk of 9615
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[7] from pop0: 8824
-> Sites to keep[7] from pop1: 8824
-> [readdata] lastread:8824 posi:20006767
-> Comparing positions: 1 with 0 has:8824
-> Will now do fst temp dump using a chunk of 8824
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[8] from pop0: 16633
-> Sites to keep[8] from pop1: 16633
-> [readdata] lastread:16633 posi:20007911
-> Comparing positions: 1 with 0 has:16633
-> Will now do fst temp dump using a chunk of 16633
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> Done reading data from chromosome will prepare next chromosome
-> hello Im the master merge part of realSFS. and I'll now do a tripple bypass to find intersect
-> 1) Will set iter according to chooseChr and start and stop, and possibly using -sites
-> Sites to keep[9] from pop0: 6247
-> Sites to keep[9] from pop1: 6247
-> [readdata] lastread:6247 posi:20000499
-> Comparing positions: 1 with 0 has:6247
-> Will now do fst temp dump using a chunk of 6247
-> Only read nSites: 0 will therefore prepare next chromosome (or exit)
-> Will now do fst temp dump using a chunk of 0
-> fst index finished with no errors!
Example runs:
realSFS fst stats CEU_YRI.fst.idx
realSFS fst stats2 CEU_YRI.fst.idx
Now we will compute the average Fst for CEU vs, YRI populations.
In [23]:
realSFS fst stats CEU_YRI.fst.idx -> Assuming idxname:CEU_YRI.fst.idx
-> Assuming .fst.gz file: CEU_YRI.fst.gz
-> FST.Unweight[nObs:158204]:0.008683 Fst.Weight:0.134346
0.008683 0.134346
The average Fst is not very informative as we do not know what loci the selection signal comes from. To figure tis out, we will have to compute Fst for CEU vs, YRI populations in a sliding window.
In [24]:
realSFS fst stats2 CEU_YRI.fst.idx -win 50 -step 10 > slidingwindow_win50_step10 -> Assuming idxname:CEU_YRI.fst.idx
-> Assuming .fst.gz file: CEU_YRI.fst.gz
-> args: tole:0.000000 nthreads:4 maxiter:100 nsites(block):0 start:(null) chr:(null) start:-1 stop:-1 fstout:(null) oldout:0 seed:-1 bootstrap:0 resample_chr:0 whichFst:0 fold:0 ref:(null) anc:(null)
win:50 step:10
nSites:11218
nSites:10005
nSites:11825
nSites:7433
nSites:4580
nSites:4269
nSites:8855
nSites:1564
nSites:6271
nSites:1406
nSites:9853
nSites:11636
nSites:7716
nSites:3014
nSites:7606
nSites:6424
nSites:3210
nSites:9615
nSites:8824
nSites:16633
nSites:6247
And finally we will plot the results using custom R scripts:
In [27]:
df <- read.delim("slidingwindow_win50_step10", header = FALSE)
df <- df[-1, ]
df$V2 <- as.numeric(df$V2)
df$V3 <- as.numeric(df$V3)
df$V5 <- as.numeric(df$V5)
df <- df[order(df$V2, df$V3), ]
rownames(df) <- seq(1, dim(df)[1], 1)
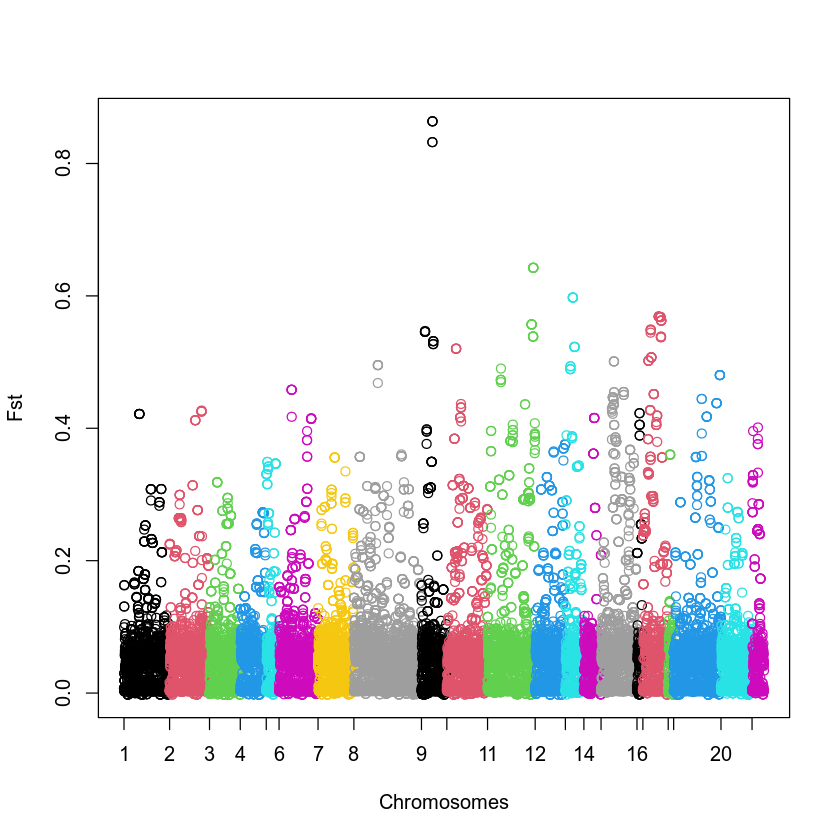
plot(df$V5, col = df$V2, xlab = "Chromosomes", ylab = "Fst", xaxt = "n")
myticks <- as.numeric(rownames(df[!duplicated(df$V2), ]))
axis(side = 1, at = myticks, labels = seq(1, 21, 1))