Assembly Validation Exercises.
Introduction
The following exercises are intended to introduce you to the tools involved in assembly validation.
Many commands below are wrapped up in functions to make it easier for you to run.
Functions can be copy-and-pasted into your terminal window or you can write them in a file
and source the file (e.g. source functions.sh).
A function looks like the following:
run_many_commands () {
ARG_1="$1" # This is the first parameter
ARG_2="$2" # This is the second parameter
ARG_3="$3" # This is the third parameter
# Then follows a complex series of commands to get a result
command_1 "$ARG_1" "$ARG_2" > new.file
command_2 new.file "$ARG_3" > result.file
}Once this command is copied into your terminal, you can use the function to run the complex series of commands:
run_many_commands File1.ext File2.ext File3.ext
# Your results will now be in result.file as written in the function aboveExercises
Some assemblies have been provided for you to assess. They can be found here:
/proj/sllstore2017027/workshop-GA2018/data/SRR492065_assembliesThis is how the assemblies were made:
| Assembly | Description |
|---|---|
| spades_k21-55_full.fasta | spades using k=21,33,55 and SRR492065_{1,2}.fastq.gz |
| spades_k21-127_full.fasta | spades using k=21,33,55,77,99,127 and SRR492065_{1,2}.fastq.gz |
| spades_k21-55_normalized.fasta | spades using k=21,33,55 and SRR492065normalized{1,2}.fastq.gz |
| spades_k21-127_normalized.fasta | spades using k=21,33,55,77,99,127 and SRR492065normalized{1,2}.fastq.gz |
| spades_k21-55_cleaned.fasta | spades using k=21,33,55 and SRR492065cleaned{1,2}.fastq.gz |
| spades_k21-127_cleaned.fasta | spades using k=21,33,55,77,99,127 and SRR492065cleaned{1,2}.fastq.gz |
| shovill_full_spades.fasta | shovill using assembler=spades and SRR492065_{1,2}.fastq.gz |
| shovill_full_megahit.fasta | shovill using assembler=megahit and SRR492065_{1,2}.fastq.gz |
| masurca_cleaned.fasta | masurca using SRR492065cleaned{1,2}.fastq.gz |
| abyss_k35_cleaned.fasta | abyss using k=35 and SRR492065cleaned{1,2}.fastq.gz |
Task 1.
QUAST is a good starting point to help evaluate the quality of assemblies. It provides many helpful contiguity statistics.
Run QUAST on all the assemblies at once and generate a report.
module load bioinfo-tools quast/4.5.4
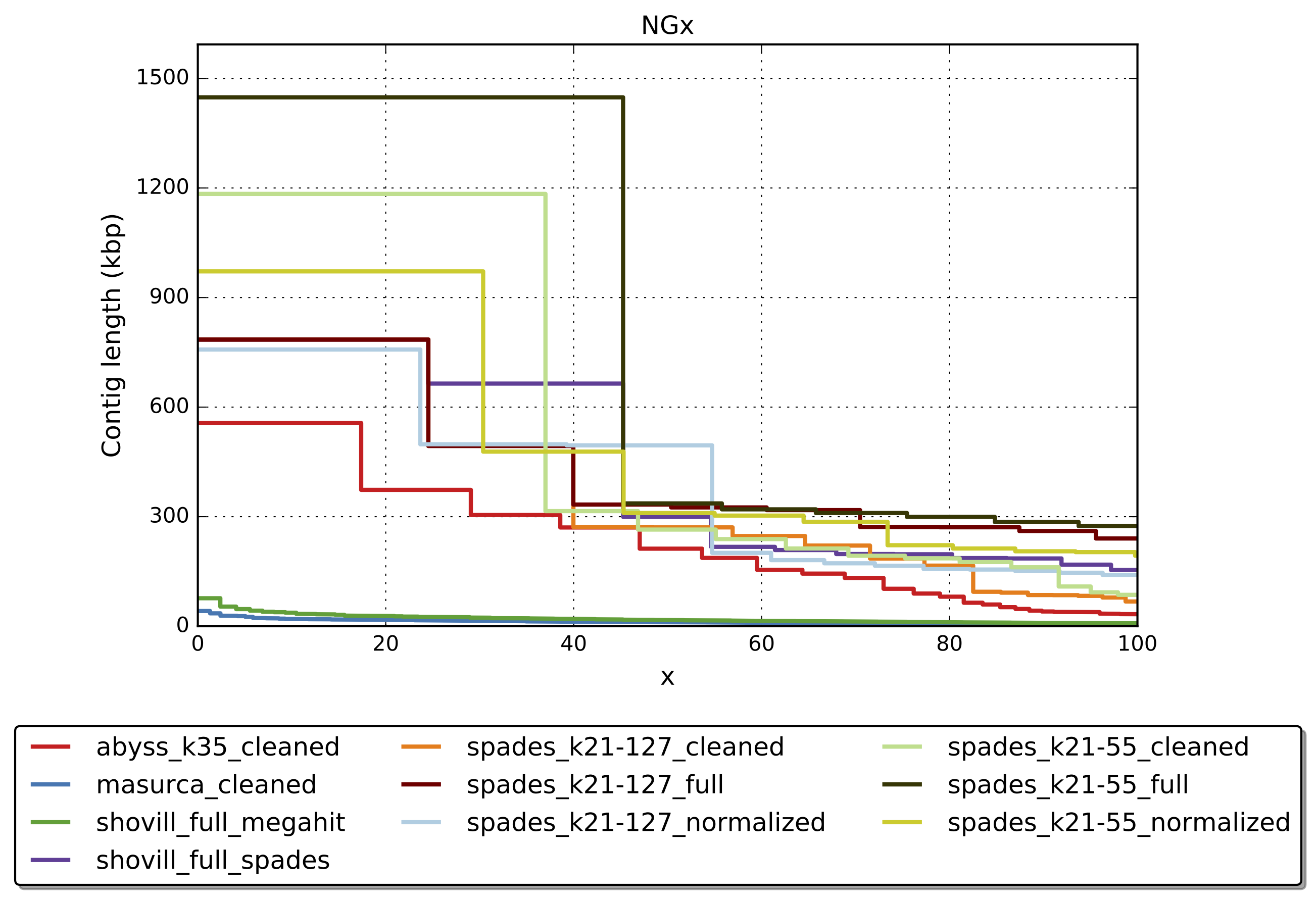
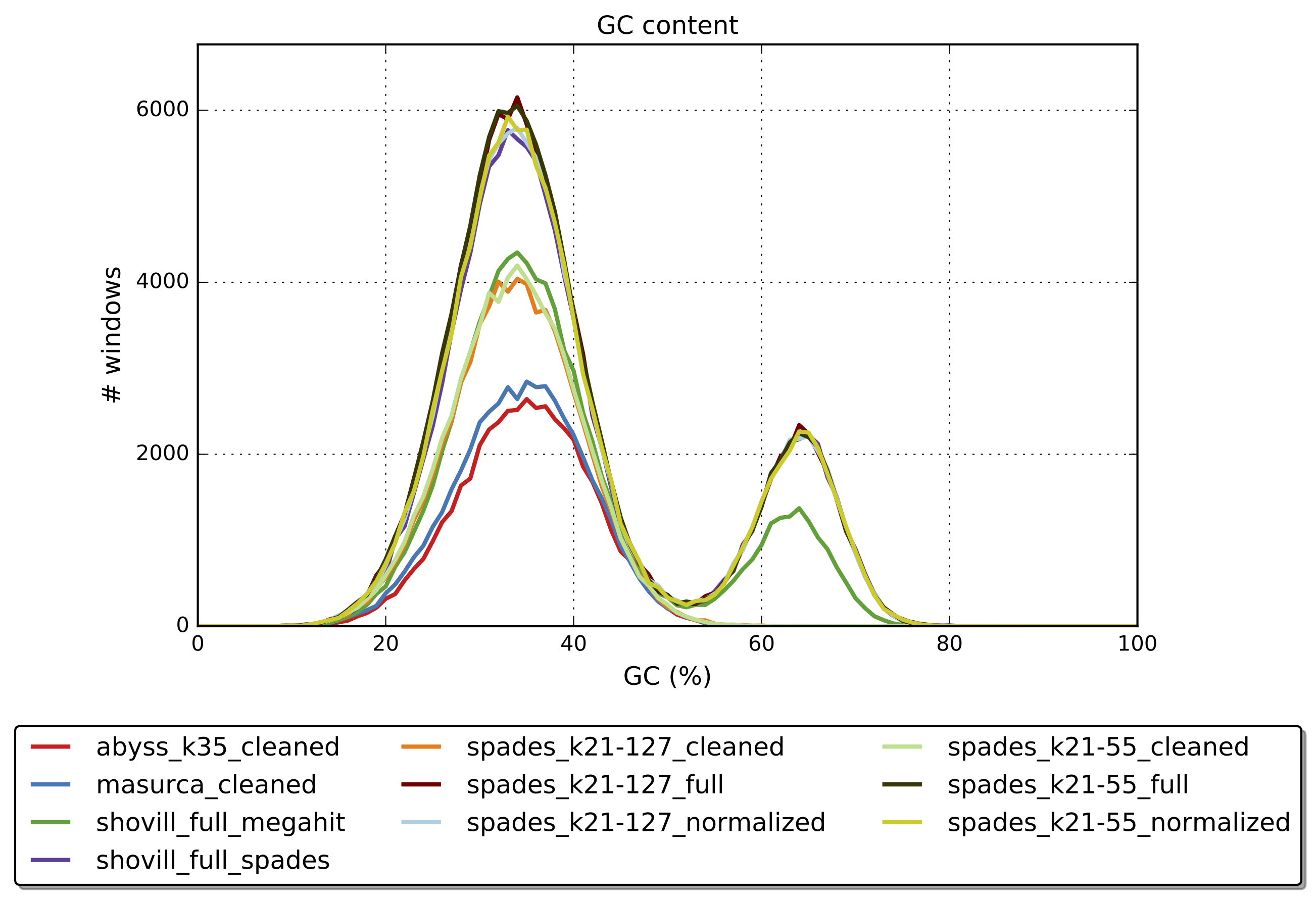
quast.py --helpWhich assembly looks the best?
Solution - click to expand
First run Quast on all the assemblies.
quast.py -t "${CPUS:-10}" --est-ref-size 3200000 *.fasta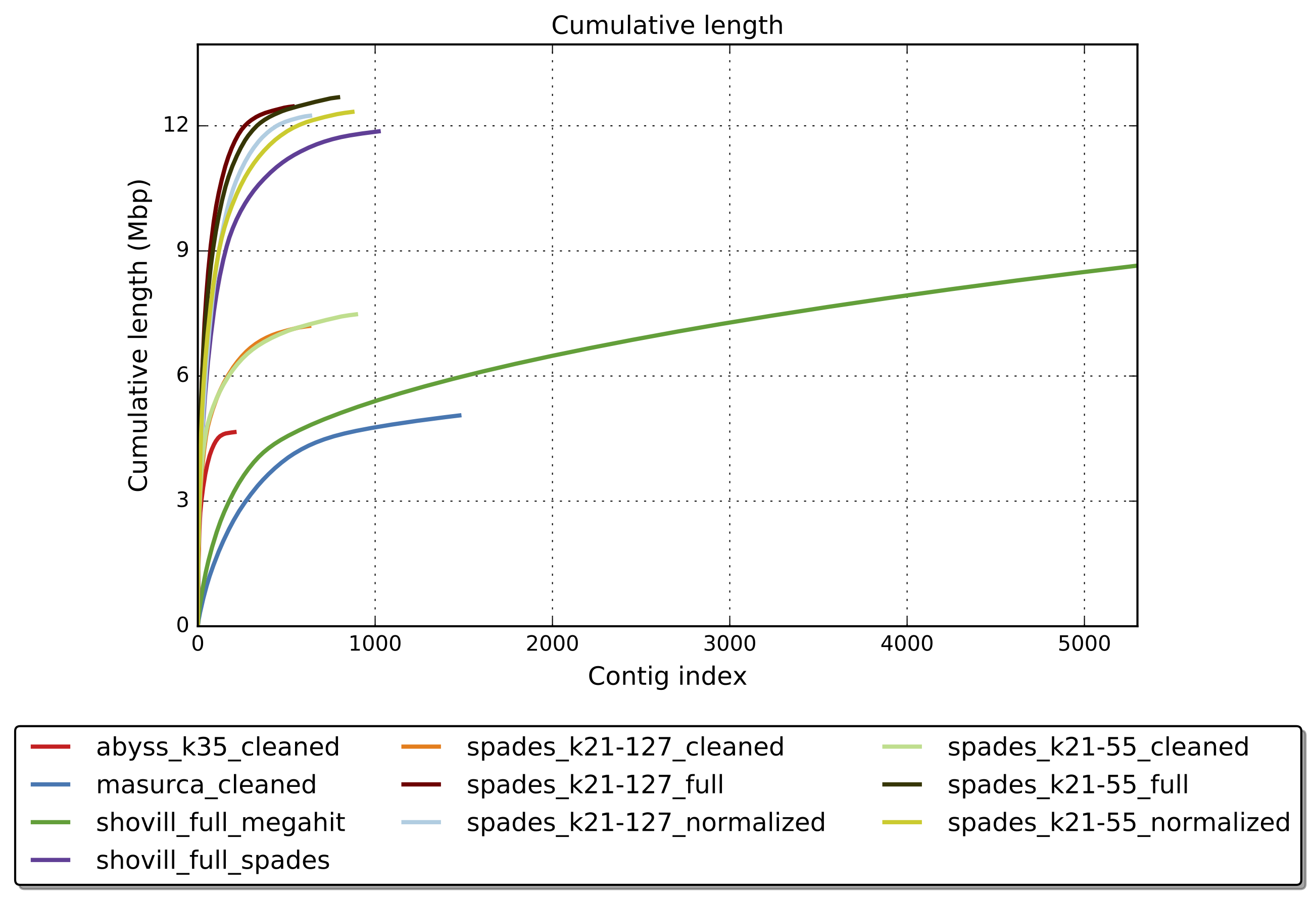
Task 2.
Select three assemblies of your choice for the remaining tasks.
Read congruency is an important measure in determining assembly accuracy. Clusters of read pairs that align incorrectly are strong indicators of mis-assembly.
How well do the reads align back to the draft assemblies? Use bwa and samtools to assess the basic alignment statistics.
Make a folder for your results.
module load bioinfo-tools bwa/0.7.17 samtools/1.8
mkdir BWA
cd BWA
ln -s ../*.fasta . # link the fasta files in this directoryThen copy this function into your terminal.
align_reads () {
ASSEMBLY="$1" # The assembly is the first parameter to this function. Must end in .fasta
READ1="$2" # The first read pair is the second parameter to this function
READ2="$3" # The second read pair is the third parameter to this function
PREFIX=$(basename "$ASSEMBLY" .fasta )
bwa index "$ASSEMBLY" # Index the assembly prior to alignment
bwa mem -t "${CPUS:-10}" "$ASSEMBLY" "$READ1" "$READ2" | samtools sort -@ "${CPUS:-10}" -T "${PREFIX}" -O BAM -o "${PREFIX}_bwa_alignment.bam" -
samtools index "${PREFIX}_bwa_alignment.bam"
samtools flagstat "${PREFIX}_bwa_alignment.bam" > "${PREFIX}_bwa_alignment.flagstats"
}To run the function above, copy the function into your terminal window and use in the following way:
align_reads assembly.fasta read1.fastq.gz read2.fastq.gzThis will then run bwa index, bwa mem, samtools sort, samtools index, and samtools flagstat in the correct
order and with the correct parameters.
Solution - click to expand
align_reads abyss_k35_cleaned.fasta ../SRR492065_cleaned_R1.fastq.gz ../SRR492065_cleaned_R2.fastq.gz
align_reads masurca_cleaned.fasta ../SRR492065_cleaned_R1.fastq.gz ../SRR492065_cleaned_R2.fastq.gz
align_reads shovill_full_megahit.fasta ../SRR492065_1.fastq.gz ../SRR492065_2.fastq.gz
align_reads shovill_full_spades.fasta ../SRR492065_1.fastq.gz ../SRR492065_2.fastq.gz
align_reads spades_k21-127_cleaned.fasta ../SRR492065_cleaned_R1.fastq.gz ../SRR492065_cleaned_R2.fastq.gz
align_reads spades_k21-127_full.fasta ../SRR492065_1.fastq.gz ../SRR492065_2.fastq.gz
align_reads spades_k21-127_normalized.fasta ../SRR492065_normalized_1.fastq.gz ../SRR492065_normalized_2.fastq.gz
align_reads spades_k21-55_cleaned.fasta ../SRR492065_cleaned_R1.fastq.gz ../SRR492065_cleaned_R2.fastq.gz
align_reads spades_k21-55_full.fasta ../SRR492065_1.fastq.gz ../SRR492065_2.fastq.gz
align_reads spades_k21-55_normalized.fasta ../SRR492065_normalized_1.fastq.gz ../SRR492065_normalized_2.fastq.gz# abyss_k35_cleaned_bwa_alignment.flagstats
8173153 + 0 mapped (95.66% : N/A)
7791892 + 0 properly paired (91.28% : N/A)
# masurca_cleaned_bwa_alignment.flagstats
7445760 + 0 mapped (87.19% : N/A)
6245750 + 0 properly paired (73.17% : N/A)
# shovill_full_megahit_bwa_alignment.flagstats
10405980 + 0 mapped (97.08% : N/A)
7204140 + 0 properly paired (67.27% : N/A)
# shovill_full_spades_bwa_alignment.flagstats
10407264 + 0 mapped (97.14% : N/A)
9776868 + 0 properly paired (91.30% : N/A)
# spades_k21-127_cleaned_bwa_alignment.flagstats
8365652 + 0 mapped (97.96% : N/A)
8040432 + 0 properly paired (94.19% : N/A)
# spades_k21-127_full_bwa_alignment.flagstats
10426814 + 0 mapped (97.32% : N/A)
9895100 + 0 properly paired (92.40% : N/A)
# spades_k21-127_normalized_bwa_alignment.flagstats
8711547 + 0 mapped (98.67% : N/A)
8335810 + 0 properly paired (94.44% : N/A)
# spades_k21-55_cleaned_bwa_alignment.flagstats
8385452 + 0 mapped (98.19% : N/A)
8061476 + 0 properly paired (94.44% : N/A)
# spades_k21-55_full_bwa_alignment.flagstats
10444190 + 0 mapped (97.48% : N/A)
9882702 + 0 properly paired (92.29% : N/A)
# spades_k21-55_normalized_bwa_alignment.flagstats
8722078 + 0 mapped (98.79% : N/A)
8328482 + 0 properly paired (94.35% : N/A)Task 3.
Use the alignments to search for signals of misassembly.
source /proj/sllstore2017027/workshop-GA2018/tools/anaconda/miniconda2/etc/profile.d/conda.sh
conda activate GA2018_frc
mkdir FRC
cd FRC
ln -s ../BWA/*.bam .
apply_FRC () {
ALIGNMENT="$1" # The BAM alignment file is the first parameter to this function. The filename must end in .bam.
GENOME_SIZE="$2" # The estimated genome size is the second parameter to this function
PREFIX=$( basename "$ALIGNMENT" .bam )
FRC --pe-sam "$ALIGNMENT" --genome-size "$GENOME_SIZE" --output "${PREFIX}"
}Plot the FRC curves (${PREFIX}_FRC.txt) together in a plot using Gnuplot. Which assembly has the best feature response curve?
gnuplot << EOF
set terminal png size 800,600
set output 'FRC_Curve_all_assemblies.png'
set title "FRC Curve" font ",14"
set key right bottom font ",8"
set autoscale
set ylabel "Approximate Coverage (%)"
set xlabel "Feature Threshold"
files = system('find -name "*alignment_FRC.txt"')
plot for [data in files] data using 1:2 with lines title data
EOFHow do these results compare to the Quast results?
Solution - click to expand
for BAM in *.bam; do
apply_FRC "$BAM" 3200000
done
gnuplot << EOF
set terminal png size 800,600
set output 'FRC_Curve_all_assemblies.png'
set title "FRC Curve" font ",14"
set key right bottom font ",8"
set autoscale
set ylabel "Approximate Coverage (%)"
set xlabel "Feature Threshold"
files = system('find -name "*alignment_FRC.txt"')
plot for [data in files] data using 1:2 with lines title data
EOF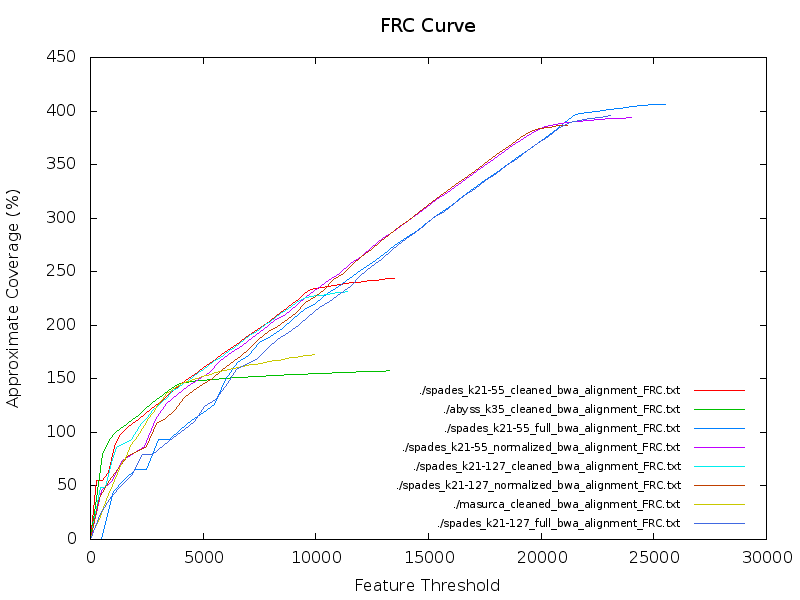
Task 4.
Use IGV to load an assembly, BAM file, and the corresponding FRC GFF file.
Note that running a graphical application over the network is a very slow process. If you have IGV installed on your computer, please download the files locally and view them there. Otherwise IGV can be started on your node using:
module load bioinfo-tools IGV/2.4.2
igv.shIn order for IGV to load the BAM file, you need to make also download the index for it as well.
Task 5.
KAT is useful tool for high accuracy sequence data. The spectra-cn (copy number spectra) graph shows a decomposition of k-mers in the assembly vs k-mers in the reads. The black portion are k-mers not present in the assembly, the red portion is found once in the assembly, and so on. This shows the completeness of an assembly, i.e. are all the reads assembled into contigs representative of the sequence data.
Use KAT to compare the assembly against the reads.
source /proj/sllstore2017027/workshop-GA2018/tools/anaconda/miniconda2/etc/profile.d/conda.sh
conda activate GA2018
mkdir KAT
cd KAT
ln -s ../*.fasta .
apply_katcomp () {
ASSEMBLY="$1" # The assembly is the first parameter to this function. It must end in .fasta
READ1="$2" # The first read pair is the second parameter to this function
READ2="$3" # The second read pair is the third parameter to this function
PREFIX=$( basename "${ASSEMBLY}" .fasta)
TMP_FASTQ=$(mktemp -u --suffix ".fastq")
mkfifo "${TMP_FASTQ}" && zcat "$READ1" "$READ2" > "${TMP_FASTQ}" & # Make a named pipe and combine reads
sleep 5 # Give a little time for the pipe to be made
kat comp -H 800000000 -t "${CPUS:-10}" -o "${PREFIX}_vs_reads.cmp" "${TMP_FASTQ}" "$ASSEMBLY" # Compare Reads to Assembly
rm "${TMP_FASTQ}"
}Solution - click to expand
apply_katcomp abyss_k35_cleaned.fasta ../SRR492065_cleaned_R1.fastq.gz ../SRR492065_cleaned_R2.fastq.gz
apply_katcomp masurca_cleaned.fasta ../SRR492065_cleaned_R1.fastq.gz ../SRR492065_cleaned_R2.fastq.gz
apply_katcomp shovill_full_megahit.fasta ../SRR492065_1.fastq.gz ../SRR492065_2.fastq.gz
apply_katcomp shovill_full_spades.fasta ../SRR492065_1.fastq.gz ../SRR492065_2.fastq.gz
apply_katcomp spades_k21-127_cleaned.fasta ../SRR492065_cleaned_R1.fastq.gz ../SRR492065_cleaned_R2.fastq.gz
apply_katcomp spades_k21-127_full.fasta ../SRR492065_1.fastq.gz ../SRR492065_2.fastq.gz
apply_katcomp spades_k21-127_normalized.fasta ../SRR492065_normalized_1.fastq.gz ../SRR492065_normalized_2.fastq.gz
apply_katcomp spades_k21-55_cleaned.fasta ../SRR492065_cleaned_R1.fastq.gz ../SRR492065_cleaned_R2.fastq.gz
apply_katcomp spades_k21-55_full.fasta ../SRR492065_1.fastq.gz ../SRR492065_2.fastq.gz
apply_katcomp spades_k21-55_normalized.fasta ../SRR492065_normalized_1.fastq.gz ../SRR492065_normalized_2.fastq.gz