Data Quality Assessment
Exercises
Task 1.
What is a k-mer?
Solution - click to expand
A sequence of characters of length k.Task 2.
How many 4-mers are in the following sequence?
ACGTTTATCCTATACGGTAATAC
Solution - click to expand
20 (L-k+1)=(23-4+1)Task 3.
What are the frequencies of the 4-mers in the sequence above.
Solution - click to expand
ACGT 1 CGTT 1 GTTT 1 TTTA 1 TTAT 1
TATC 1 ATCC 1 TCCT 1 CCTA 1 CTAT 1
TATA 1 ATAC 2 TACG 1 ACGG 1 CGGT 1
GGTA 1 GTAA 1 TAAT 1 AATA 1Task 4.
Load Kat with the following command:
source /proj/sllstore2017027/workshop-GA2018/tools/anaconda/miniconda2/etc/profile.d/conda.sh
conda activate GA2018
kat --helpUse kat hist to obtain a histogram of data Enterococcus_faecalis/SRR492065_{1,2}.fastq.gz.
What is the approximate k-mer coverage? Is anything wrong with this histogram?
# Make a named pipe and run in the background
TMPFILE=$(mktemp -u --suffix ".fastq")
mkfifo "$TMPFILE" && zcat "$READ1" "$READ2" > "$TMPFILE" &
# Run KAT reading from the named pipe
kat hist -t "$CPUS" -o "${PREFIX}.hist" "$TMPFILE"
rm "$TMPFILE"Solution - click to expand
CPUS=${SLURM_NPROCS:-10}
READ1=Enterococcus_faecalis/SRR492065_1.fastq.gz
READ2=Enterococcus_faecalis/SRR492065_2.fastq.gz
PREFIX=$(basename ${READ1%_1.*} )
TMPFILE=$(mktemp -u --suffix ".fastq")
mkfifo "$TMPFILE" && zcat "$READ1" "$READ2" > "$TMPFILE" &
kat hist -t "$CPUS" -o "${PREFIX}.hist" "$TMPFILE"
rm "$TMPFILE"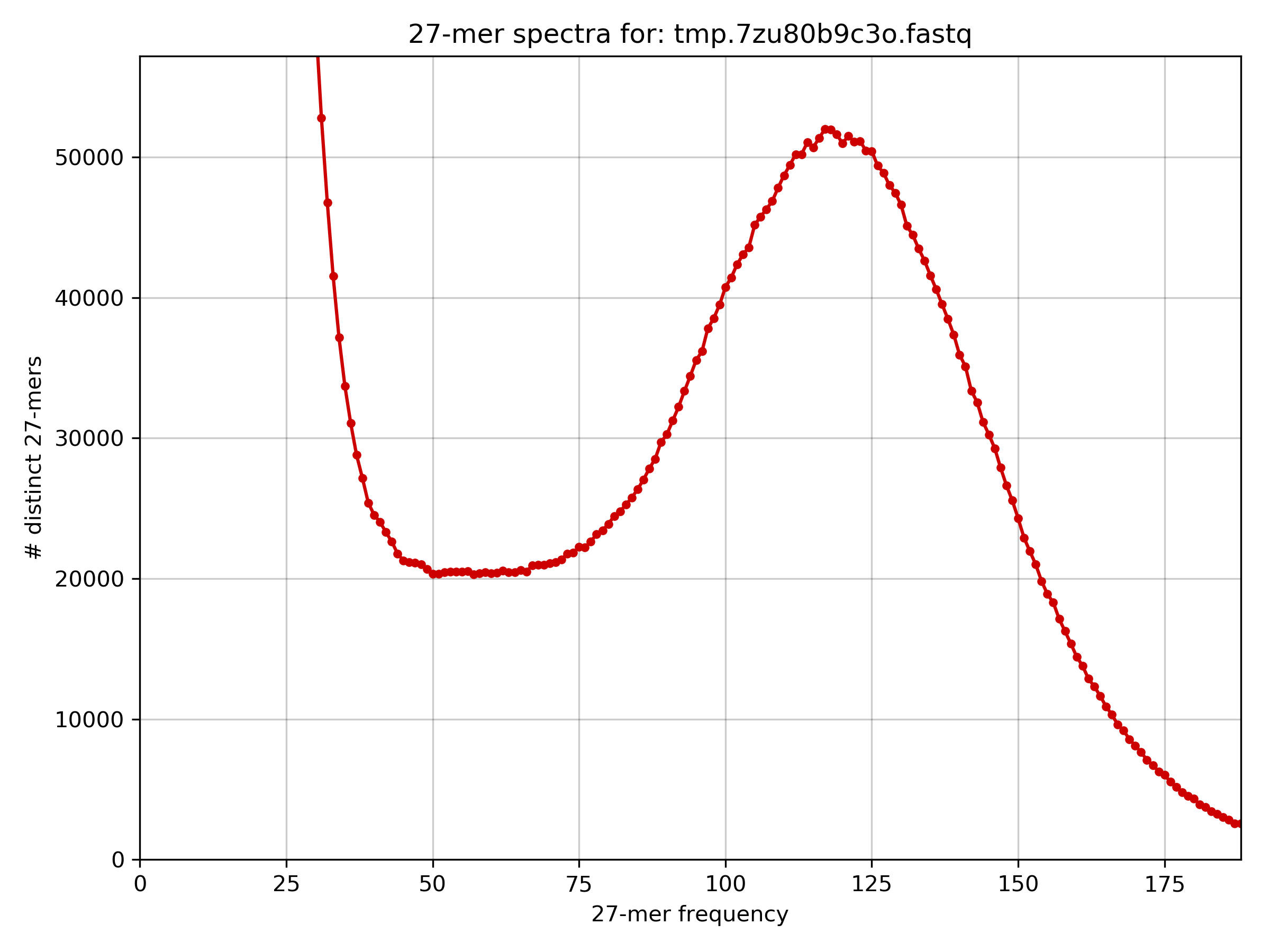
The homozygous peak in the histogram is at 116x k-mer coverage.
The histogram is unusual because there is a higher than expected frequency of low frequency k-mers.
Task 5.
Use the histogram to estimate the genome size from the homozygous peak. Does this correspond to the estimated genome size of Enterococcus faecalis?
Solution - click to expand
The peak of the histogram is at around 116x coverage, read length is 100, and the k-mer size is 27. This leads to a read depth of M * L / (L - K + 1) = 116 * 100 / (100 - 27 + 1) =~ 156x read depth of coverage. The total number of bases in the data set is 1070871200, and therefore the estimated genome size is G = T / N = 1070871200 / 156 = 6864559 =~ 6.8 Mb. From before we know the genome size is 3.22 Mb, so the estimated genome size also indicates something unusual about the data.Task 6.
Use kat gcp to plot the k-mer frequency vs gc content.
Why is the Y-axis from 0-27, and how can you estimate the average GC% of the blobs from this scale?
# Make a named pipe and run in the background
TMPFILE=$(mktemp -u --suffix ".fastq")
mkfifo "$TMPFILE" && zcat "$READ1" "$READ2" > "$TMPFILE" &
# Run KAT reading from the named pipe
kat gcp -t "$CPUS" -o "${PREFIX}.gcp" "$TMPFILE"
rm "$TMPFILE"Solution - click to expand
READ1=Enterococcus_faecalis/SRR492065_1.fastq.gz
READ2=Enterococcus_faecalis/SRR492065_2.fastq.gz
PREFIX=$(basename ${READ1%_1.*} )
TMPFILE=$(mktemp -u --suffix ".fastq")
mkfifo "$TMPFILE" && zcat "$READ1" "$READ2" > "$TMPFILE" &
kat gcp -t "$CPUS" -o "${PREFIX}.gcp" "$TMPFILE"
rm "$TMPFILE"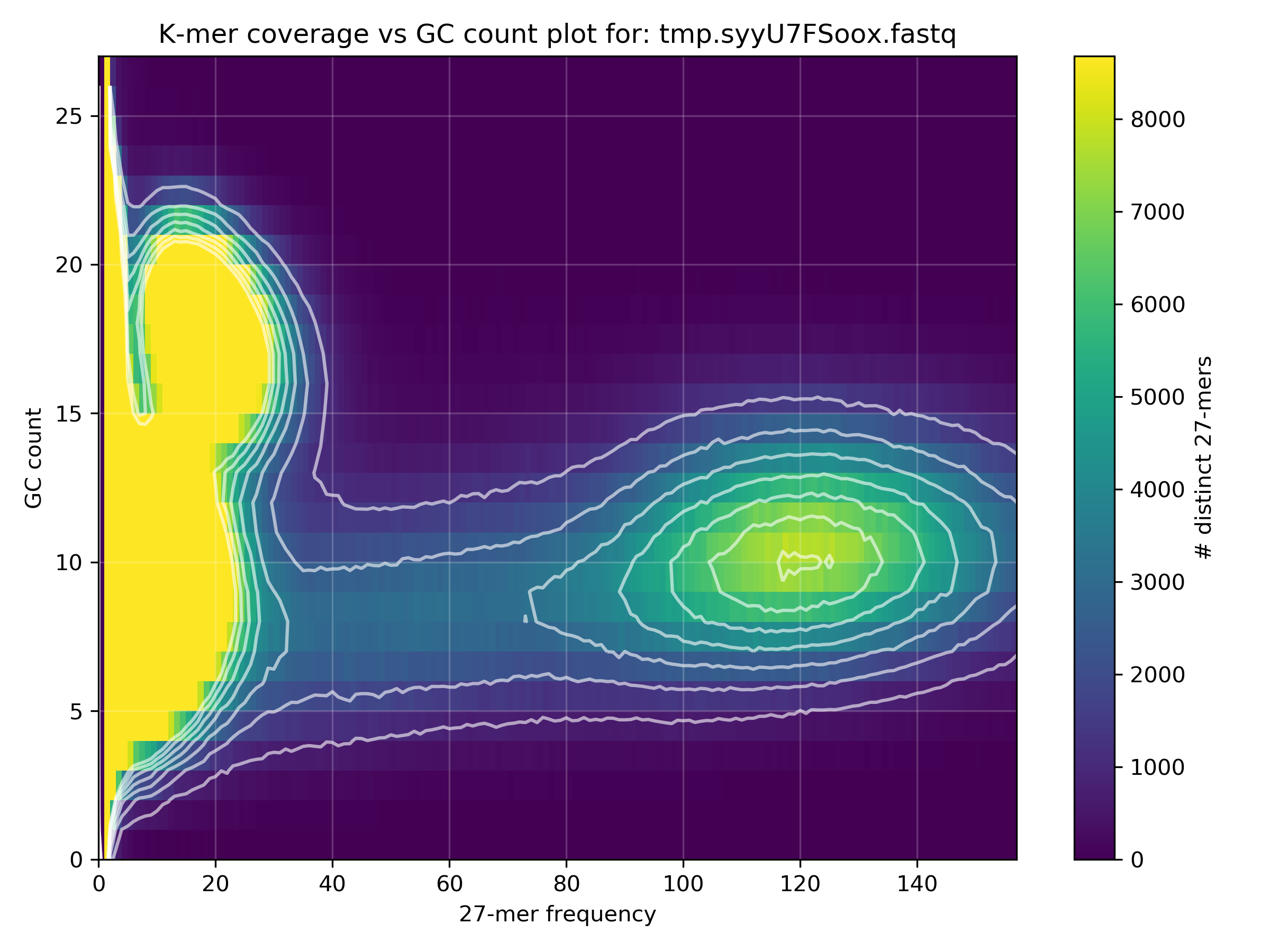
The GC content scale (Y-axis) is the absolute GC count per k-mer. The default k-mer size is 27 and therefore the y-axis is from 0 to 27. One can estimate the average GC% of a blob from this scale by multiplying the approximate GC content value by 4 (since 4 * 27 == 108 =~ 100).
Task 7.
Use kat comp to compare Enterococcus_faecalis/SRR492065_{1,2}.fastq.gz.
TMPFILE1=$(mktemp -u --suffix ".fastq")
TMPFILE2=$(mktemp -u --suffix ".fastq")
mkfifo "$TMPFILE1" && zcat "$READ1" > "$TMPFILE1" &
mkfifo "$TMPFILE2" && zcat "$READ2" > "$TMPFILE2" &
kat comp -t "$CPUS" -o "${PREFIX}_r1vr2.cmp" --density_plot "$TMPFILE1" "$TMPFILE2"
kat plot spectra-mx -x 50 -y 500000 --intersection -o "${PREFIX}_r1vr2.cmp-main.mx.spectra-mx.png" "${PREFIX}_r1vr2.cmp-main.mx"
rm "$TMPFILE1" "$TMPFILE2"Why is there a difference in the distribution means between the two datasets?
Solution - click to expand
READ1=Enterococcus_faecalis/SRR492065_1.fastq.gz
READ2=Enterococcus_faecalis/SRR492065_2.fastq.gz
PREFIX=$(basename ${READ1%_1.*} )
TMPFILE1=$(mktemp -u --suffix ".fastq")
TMPFILE2=$(mktemp -u --suffix ".fastq")
mkfifo "$TMPFILE1" && zcat "$READ1" > "$TMPFILE1" &
mkfifo "$TMPFILE2" && zcat "$READ2" > "$TMPFILE2" &
kat comp -t "$CPUS" -o "${PREFIX}_r1vr2.cmp" --density_plot "$TMPFILE1" "$TMPFILE2"
kat plot spectra-mx -x 50 -y 500000 --intersection -o "${PREFIX}_r1vr2.cmp-main.mx.spectra-mx.png" "${PREFIX}_r1vr2.cmp-main.mx"
rm "$TMPFILE1" "$TMPFILE2"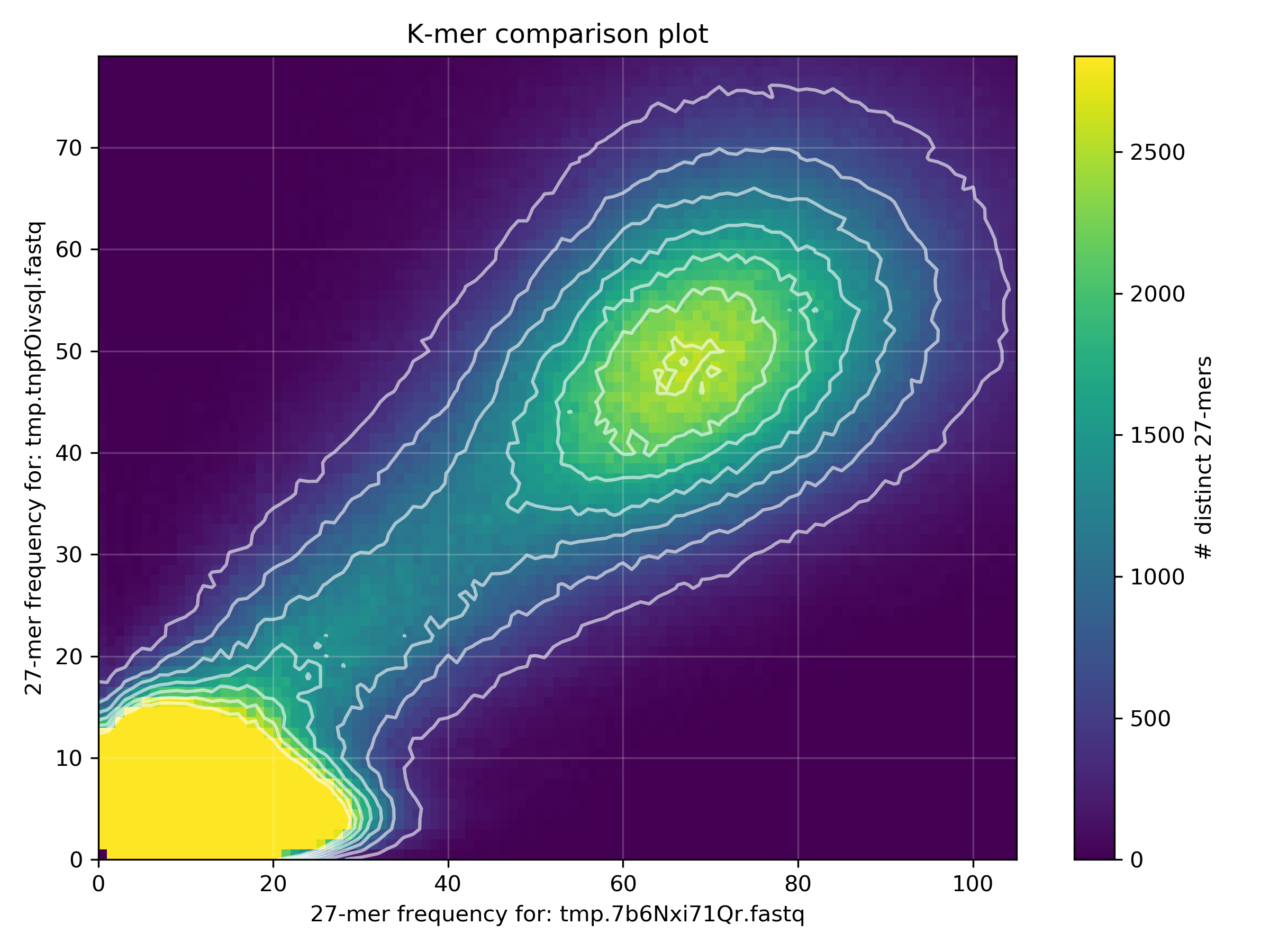

The spectra-mx plot shows the shared content for dataset 2 (READ2) is shifted to the left and slightly higher than the shared content of dataset 1 (READ1). From the previous FastQC analyses of these files we saw that READ2 has lower read qualities than READ1. Lower quality reads mean more errors and ambiguous bases, resulting in lower k-mer frequency counts (in READ2) which shifts the mean k-mer frequency to the left.