This vignette demonstrates the use of easyshiny package to create shiny web app from single-cell transcriptomic data.
A small dataset
In this example, we will use the built-in data that comes with Seurat.
pbmc_smallAn object of class Seurat
230 features across 80 samples within 1 assay
Active assay: RNA (230 features, 20 variable features)
2 dimensional reductions calculated: pca, tsneSince it has been already processed (normalisation, scaling, PCA, UMAP), we can proceed to building the app.
pbmc_conf <- create_config(pbmc_small)
make_app(pbmc_small, pbmc_conf, gex.assay = "RNA", gex.slot = "data", shiny.title = "PBMC", shiny.dir = "pbmc")This creates a directory called pbmc which contains the following files:
list.files("pbmc")pbmc
├── about.md
├── sc1conf.rds
├── sc1def.rds
├── sc1gene.rds
├── sc1gexpr.h5
├── sc1meta.rds
├── server.R
├── ui.R
└── www
└── styles.cssTo launch the app, run:
shiny::runApp("pbmc")
Use
Rscript -e 'shiny::runApp("app",launch.browser=F)' if
running from the terminal.
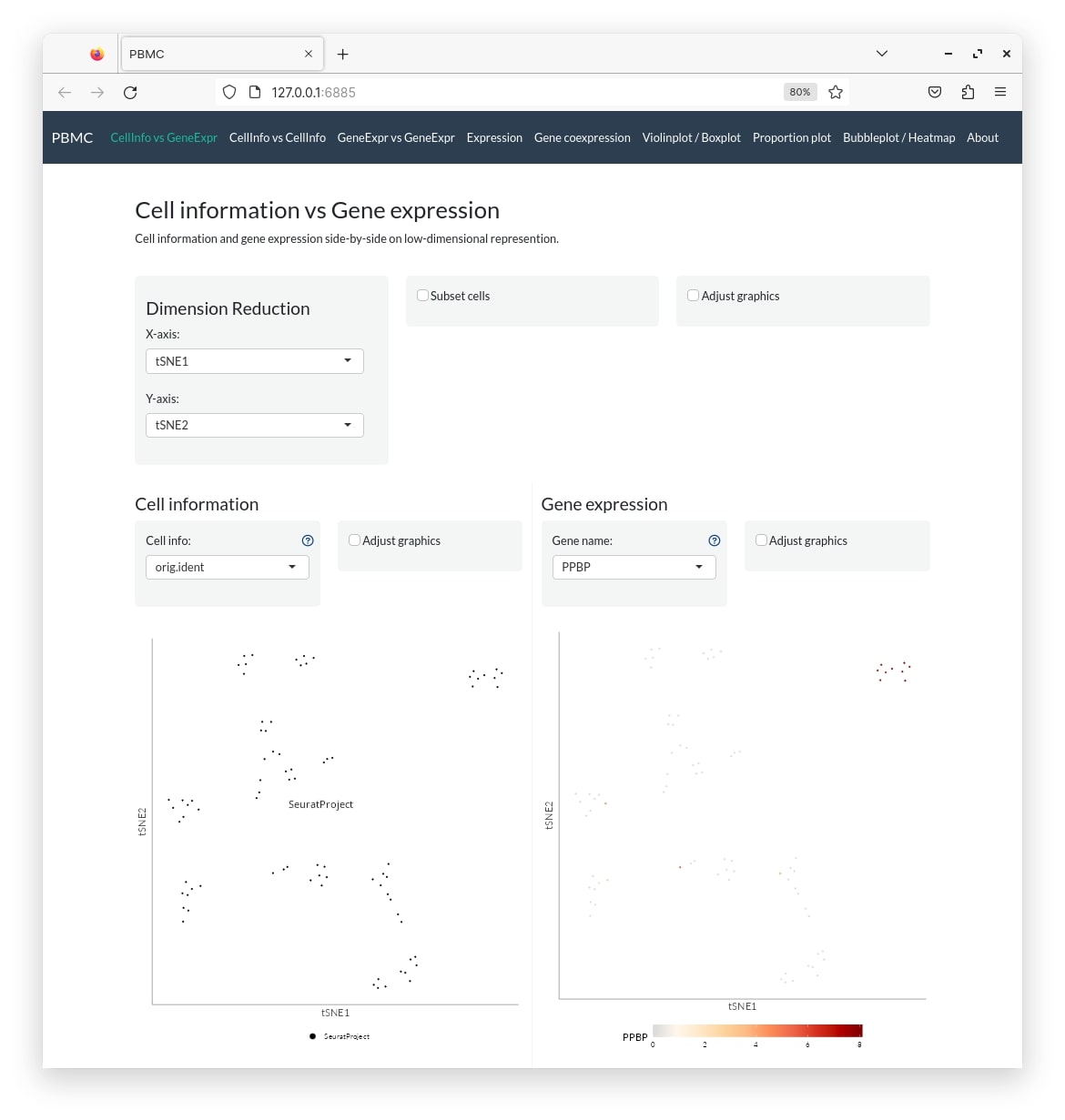
A larger dataset
We will now try a larger dataset using the SeuratData package.
library(SeuratData)
obj <- LoadData("panc8")
objAn object of class Seurat
34363 features across 14890 samples within 1 assay
Active assay: RNA (34363 features, 0 variable features)Run basic processing including normalisation, scaling, PCA and UMAP.
obj <- obj |>
NormalizeData() |>
ScaleData() |>
FindVariableFeatures() |>
RunPCA() |>
RunUMAP(dims = 1:20)
panc8_conf <- create_config(obj)
make_app(obj, panc8_conf, gex.assay = "RNA", gex.slot = "data", shiny.title = "Panc8", shiny.dir = "panc8")
shiny::runApp("panc8")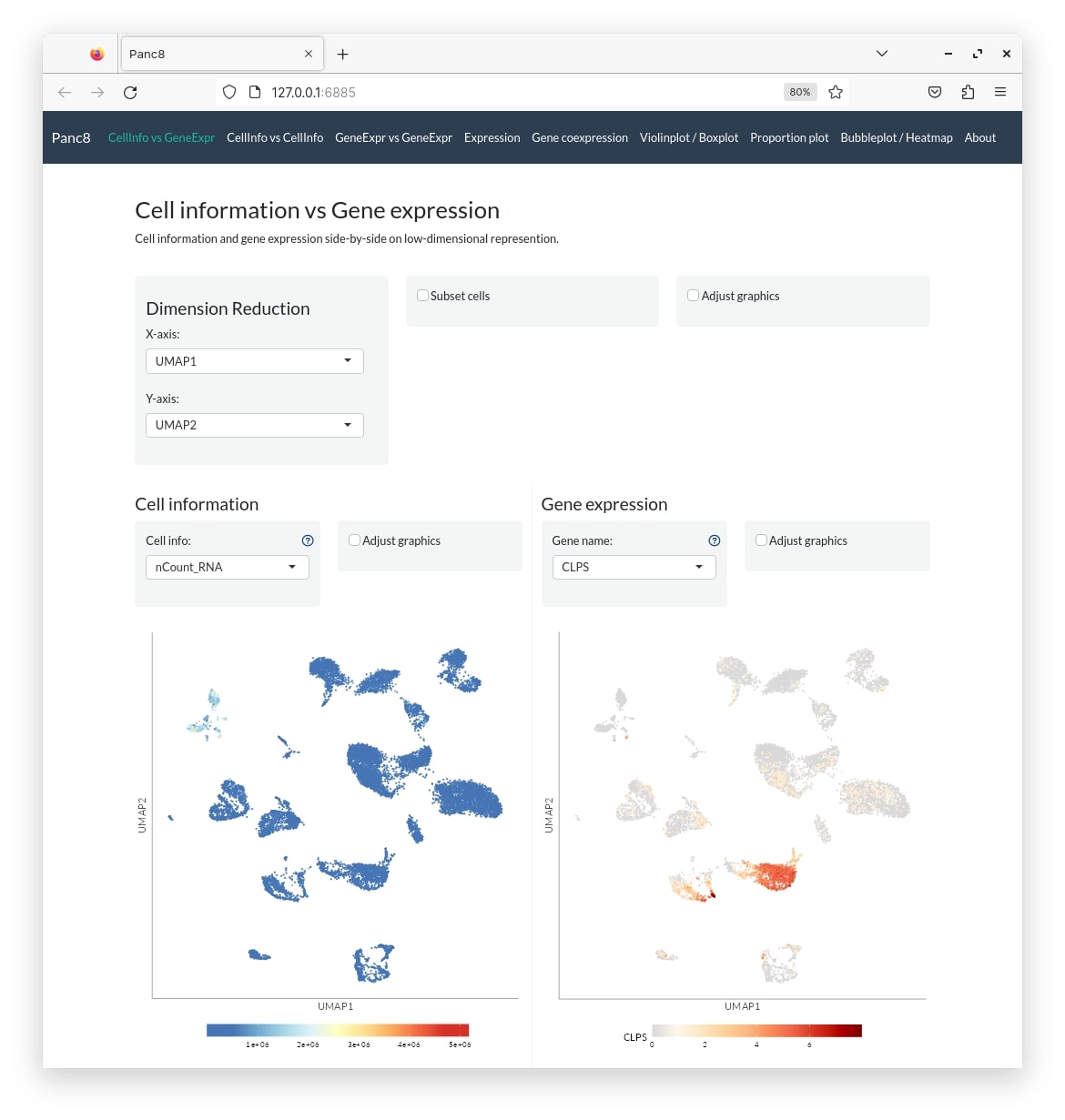
Multiple datasets
Now we will look at an example using multiple datasets. We will use the same two datasets used above. Make sure both datasets have been pre-processed with UMAP.
pbmc_conf <- create_config(pbmc_small)
make_file(pbmc_small, pbmc_conf, gex.assay="RNA", gex.slot="data", shiny.prefix = "sc1", shiny.dir="app")
panc8_conf <- create_config(obj)
make_file(obj, panc8_conf, gex.assay="RNA", gex.slot="data", shiny.prefix = "sc2", shiny.dir="app")
make_code_multi(
shiny.title = "Multi-data",
shiny.prefix = c("sc1", "sc2"),
shiny.headers = c("PBMC", "Panc8"),
shiny.dir = "app")This will create a directory named app with the following files corresponding to both datasets:
app
├── about.md
├── sc1conf.rds
├── sc1def.rds
├── sc1gene.rds
├── sc1gexpr.h5
├── sc1meta.rds
├── sc2conf.rds
├── sc2def.rds
├── sc2gene.rds
├── sc2gexpr.h5
├── sc2meta.rds
├── server.R
├── ui.R
└── www
└── styles.cssLaunch the app.
shiny::runApp("app")
Preview showing mutiple datasets.
Options
We will take a look at some of the customisation options.
Selective tabs
There are 9 different tabs that display different kinds of information. These are listed below:
| Tab | Description |
|---|---|
| civge | Cell info vs gene expression |
| civci | Cell info vs cell info |
| gevge | gene expression vs gene expression |
| gem | Gene expression of multiple genes |
| gec | Gene co-expression |
| vio | Violin plot |
| pro | Proportion plot |
| hea | Heatmap / Bubble / Dot plot |
| mar | Marker gene tables |
| about | About page and additional info |
Of these, 8 tabs are displayed by default. Marker genes are not
displayed by default. More on that below. Specific tabs can be displayed
by setting the tabs argument. Tabs are displayed in the
order in which they are provided.
pbmc_conf <- create_config(pbmc_small)
make_app(pbmc_small, pbmc_conf, gex.assay="RNA", gex.slot="data",
shiny.title = "PBMC", shiny.dir="pbmc",
tabs=c("civge","gem"))
shiny::runApp("pbmc")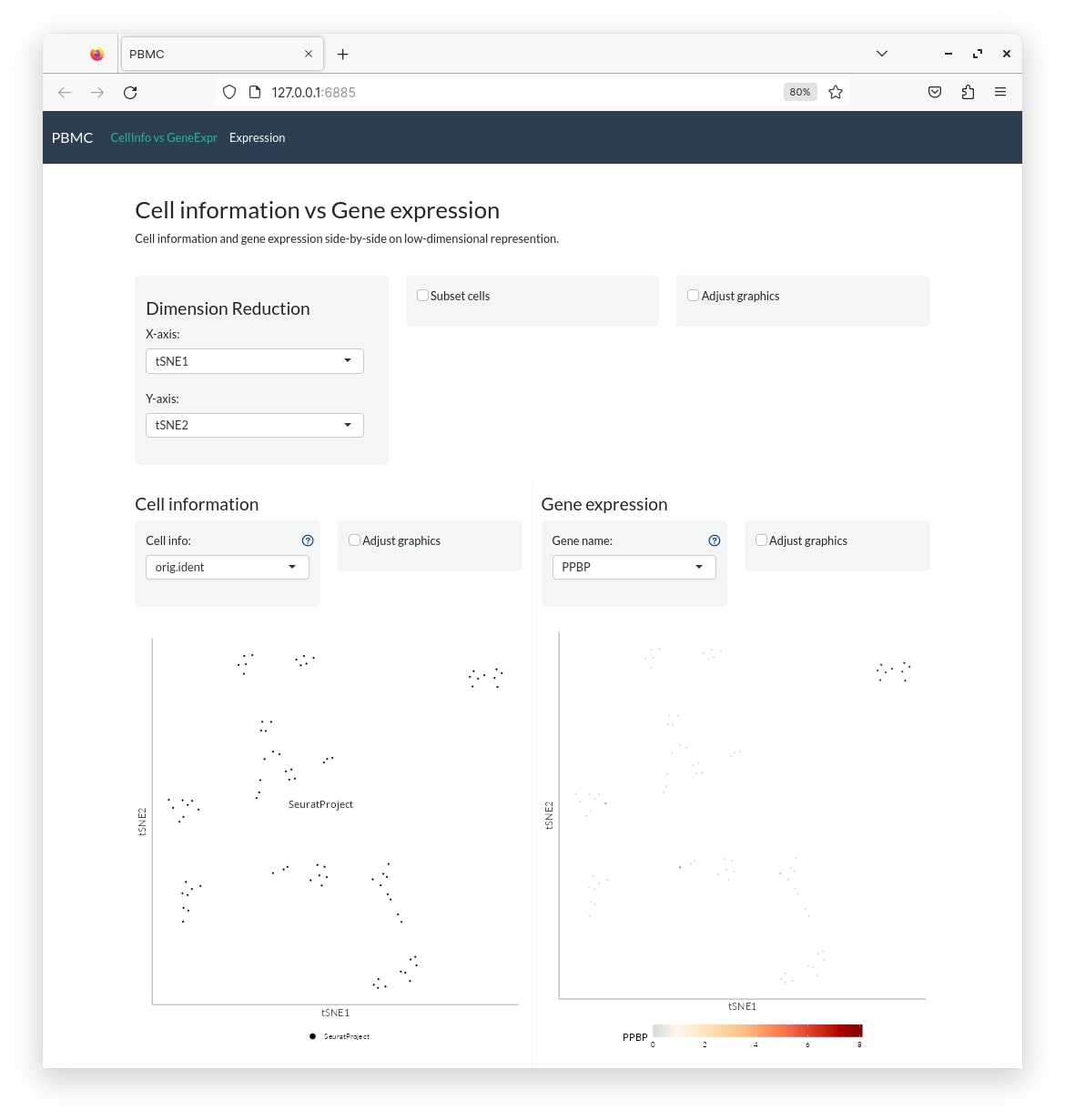
Only displaying civge and gem tabs.
About page
The about page is a tab that is displayed by
default. It contains acknowledgements by default and the user can modify
it as needed. Markdown formatting is supported. The
about page can be disabled by removing it from
tabs. When multiple datasets are used, a common
about page is created.
# app without about page
pbmc_conf <- create_config(pbmc_small)
make_app(pbmc_small, pbmc_conf, gex.assay="RNA", gex.slot="data", shiny.title = "PBMC", shiny.dir="app", tabs=c("hea","vio"))
shiny::runApp("app")Marker genes
Marker gene functionality is only implemented for Seurat objects for
now. To display marker genes, Seurat::FindAllMarkers() need
to be run and resulting table added to the Seurat object slot
misc as a named list. To simplify this, one can use this
function:
#' @description Find all markers by identity
#' @param obj Seurat v4 object
#' @param ids Column name to used as identity (one or more)
#' @param ... All arguments passed to Seurat::FindAllMarkers()
#'
fam <- function(obj,ids=NULL,...){
if(missing(obj)) stop("Seurat object not specified.")
if(is.null(ids)) stop("Argument 'ids' is NULL. It must be one or more column names from metadata.")
if(!is.character(ids)) stop("Argument 'ids' must be a character.")
if(!"Seurat" %in% class(obj)) stop("Argument 'obj' must be a Seurat V4 object.")
if(any(!ids %in% colnames(obj@meta.data))) {
stop(paste("Following ids not found in metadata:",paste(ids[!ids %in% colnames(obj@meta.data)],collapse=",")))
}
fn <- function(id=NULL,obj,...){
if(!is.null(id)) {
if(!id %in% colnames(obj@meta.data)) stop(paste0("Following column does not exist: ",id,"."))
Idents(obj) <- obj@meta.data[,id]
}
return(FindAllMarkers(obj,...))
}
return(setNames(lapply(ids,fn,obj=obj,...),ids))
}One can compute marker genes for one or more metadata columns and
resulting named list of marker tables are stored in
seurat@misc$markers.
# find marker genes for all levels in metadata column "letter.idents"
pbmc_small@misc$markers <- fam(pbmc_small,"letter.idents")
pbmc_conf <- create_config(pbmc_small)
make_app(pbmc_small, pbmc_conf, gex.assay="RNA", gex.slot="data",
shiny.title = "PBMC", shiny.dir="pbmc",
tabs=c("mar"))
shiny::runApp("pbmc")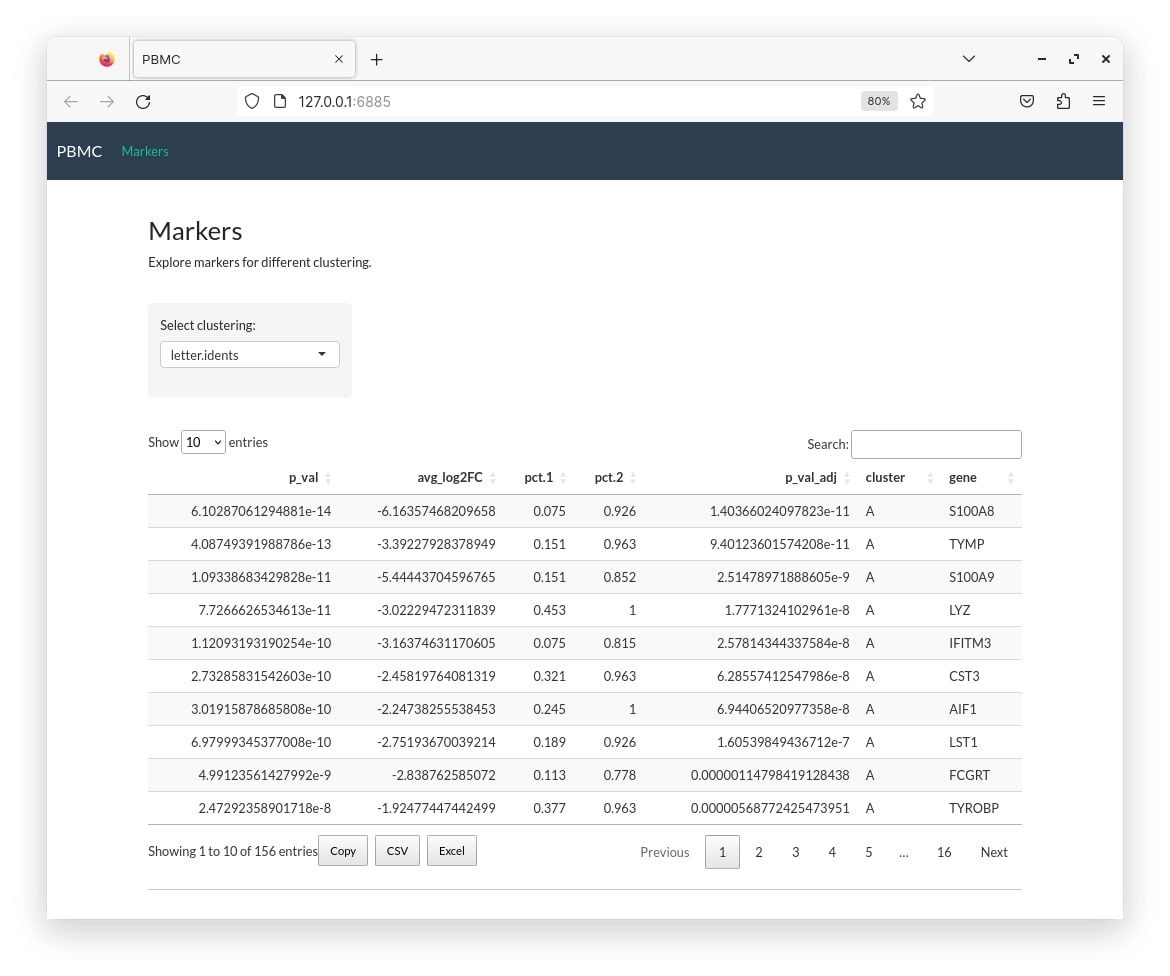
When using markers with multiple datasets, it is necessary to set
mar=TRUE in make_file().
pbmc_small@misc$markers <- fam(pbmc_small, "letter.idents")
pbmc_conf1 <- create_config(pbmc_small)
make_file(pbmc_small, pbmc_conf1, gex.assay = "RNA", gex.slot = "data", shiny.prefix = "sc1", shiny.dir = "app", mar = TRUE)
pbmc_conf2 <- create_config(pbmc_small)
make_file(pbmc_small, pbmc_conf2, gex.assay = "RNA", gex.slot = "data", shiny.prefix = "sc2", shiny.dir = "app", mar = TRUE)
make_code_multi(
shiny.title = "Multi-data",
shiny.prefix = c("sc1", "sc2"),
shiny.headers = c("PBMC 1", "PBMC 2"),
shiny.dir = "app",
tabs = c("civge", "civci", "gevge", "gem", "gec", "vio", "pro", "hea", "mar", "about"))
shiny::runApp("app")Theme
Change the app theme to another bootswatch theme. by changing the
argument theme. For more finer control over styling, edit
the www/styles.css file after creating the app.
pbmc_conf <- create_config(pbmc_small)
make_app(pbmc_small, pbmc_conf, gex.assay="RNA", gex.slot="data",
shiny.title = "PBMC", shiny.dir="pbmc", theme="united")
shiny::runApp("pbmc")
Theme changed to ‘united’.
Session
## R version 4.2.2 (2022-10-31)
## Platform: x86_64-conda-linux-gnu (64-bit)
## Running under: Zorin OS 16.2
##
## Matrix products: default
## BLAS/LAPACK: /home/roy/miniconda3/envs/r-4.2/lib/libopenblasp-r0.3.21.so
##
## locale:
## [1] LC_CTYPE=en_GB.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_GB.UTF-8 LC_COLLATE=en_GB.UTF-8
## [5] LC_MONETARY=en_GB.UTF-8 LC_MESSAGES=en_GB.UTF-8
## [7] LC_PAPER=en_GB.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_GB.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] SeuratObject_4.1.3 Seurat_4.3.0 easyshiny_0.3 shiny_1.7.4
## [5] ggplot2_3.4.1 reticulate_1.28 hdf5r_1.3.8 data.table_1.14.8
##
## loaded via a namespace (and not attached):
## [1] Rtsne_0.16 colorspace_2.1-0 deldir_1.0-6
## [4] ellipsis_0.3.2 ggridges_0.5.4 rprojroot_2.0.3
## [7] fs_1.6.1 spatstat.data_3.0-0 leiden_0.4.3
## [10] listenv_0.9.0 ggrepel_0.9.3 bit64_4.0.5
## [13] fansi_1.0.4 codetools_0.2-19 splines_4.2.2
## [16] cachem_1.0.7 knitr_1.42 polyclip_1.10-4
## [19] jsonlite_1.8.4 ica_1.0-3 cluster_2.1.4
## [22] png_0.1-8 uwot_0.1.14 spatstat.sparse_3.0-0
## [25] sctransform_0.3.5 readr_2.1.4 compiler_4.2.2
## [28] httr_1.4.4 Matrix_1.5-3 fastmap_1.1.1
## [31] lazyeval_0.2.2 cli_3.6.1 later_1.3.0
## [34] htmltools_0.5.5 tools_4.2.2 igraph_1.4.0
## [37] gtable_0.3.3 glue_1.6.2 reshape2_1.4.4
## [40] RANN_2.6.1 dplyr_1.1.1 Rcpp_1.0.10
## [43] scattermore_0.8 jquerylib_0.1.4 pkgdown_2.0.7
## [46] vctrs_0.6.1 nlme_3.1-162 spatstat.explore_3.0-6
## [49] progressr_0.13.0 lmtest_0.9-40 spatstat.random_3.1-3
## [52] xfun_0.37 stringr_1.5.0 globals_0.16.2
## [55] mime_0.12 miniUI_0.1.1.1 lifecycle_1.0.3
## [58] irlba_2.3.5.1 goftest_1.2-3 future_1.32.0
## [61] MASS_7.3-58.2 zoo_1.8-11 scales_1.2.1
## [64] spatstat.utils_3.0-1 ragg_1.2.5 hms_1.1.2
## [67] promises_1.2.0.1 parallel_4.2.2 RColorBrewer_1.1-3
## [70] yaml_2.3.7 gridExtra_2.3 memoise_2.0.1
## [73] pbapply_1.7-0 yulab.utils_0.0.6 sass_0.4.5
## [76] stringi_1.7.12 desc_1.4.2 rlang_1.1.0
## [79] pkgconfig_2.0.3 systemfonts_1.0.4 matrixStats_0.63.0
## [82] evaluate_0.20 lattice_0.20-45 tensor_1.5
## [85] ROCR_1.0-11 purrr_1.0.1 patchwork_1.1.2
## [88] htmlwidgets_1.6.1 cowplot_1.1.1 bit_4.0.5
## [91] tidyselect_1.2.0 parallelly_1.35.0 RcppAnnoy_0.0.20
## [94] plyr_1.8.8 magrittr_2.0.3 R6_2.5.1
## [97] generics_0.1.3 pillar_1.9.0 withr_2.5.0
## [100] fitdistrplus_1.1-8 abind_1.4-5 survival_3.5-3
## [103] sp_1.6-0 tibble_3.2.1 future.apply_1.10.0
## [106] KernSmooth_2.23-20 utf8_1.2.3 spatstat.geom_3.0-6
## [109] plotly_4.10.1 tzdb_0.3.0 rmarkdown_2.20
## [112] grid_4.2.2 digest_0.6.31 xtable_1.8-4
## [115] tidyr_1.3.0 httpuv_1.6.9 gridGraphics_0.5-1
## [118] textshaping_0.3.6 munsell_0.5.0 viridisLite_0.4.1
## [121] ggplotify_0.1.0 bslib_0.4.2.9000