Precourse
This workshop is aimed towards biologists, researchers, computer scientists or data analysts planning to run, analyse and interpret an RNA-Seq experiment.
On this page, you will find information and tasks for you should do to prepare for the course. Please try to do them as soon as possible, so we have time before the course starts to fix installations and other problems you might have.
Briefly, these entitle to:
- Refresh your memory in basics of Linux and R (requirements for the workshop)
- Install the software and packages required for the course using Conda
- Download the Data used during the course practicals
- Download the Code from the course
- Create your account on Canvas
Feel free to contact us if you encounter any issue. Once you have joined the course in Canvas, you can do so via the Discussions section entitled “installation and pre-course related issues”. For any issue related to creating your account on Canvas, please email us at edu.rnaseq [at] nbis.se. We have also created an FAQ with solutions to issues from previous course participants (access it by clicking “Next” as the bottom of this page), please have a look as you might encounter the same.
Requirements
![]()
We strongly recommend for those not yet familiar with UNIX and/or R to take this opportunity and take these online tutorials, since those are requirements for the workshop. This will help you to develop your programming skills and we can always learn a few tricks here and there, even if you are already experienced.
- UNIX (part_1): http://swcarpentry.github.io/shell-novice/
- UNIX (part_2): https://carpentries-incubator.github.io/shell-extras/
- R (part_1): https://swcarpentry.github.io/r-novice-inflammation/
- R (part_2): http://swcarpentry.github.io/r-novice-gapminder/
After taking those courses (or any other equivalent course in programming in bash and R) will provide you with the basics in, for example:
- file structure and manipulation in bash and R
- loading, handling and manipulating vectors, matrices, factors and lists in R
- creating for-loops in R and bash
- using Rmarkdown for reports in R
- editing and writing files in the command line and in R
- and much more …
Conda

During this workshop, you will use conda environments to run the exercises. This is because conda environments allow all users to have the same computing environment, i.e. package versions. This enforces reproducibility for you to run this material without the need to re-install or change your local versions. See a graphical example below:
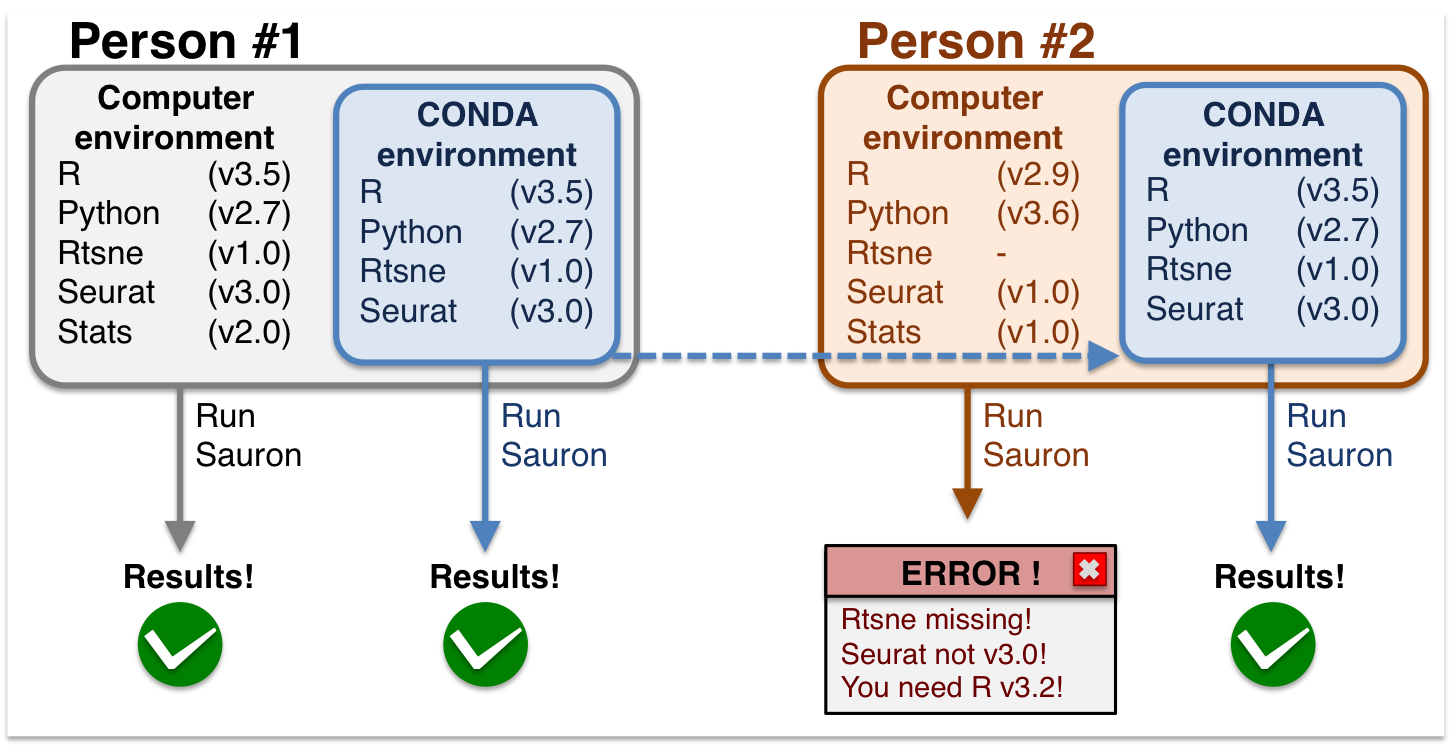
Conda environments are a self-contained directory that you can use in order to reproduce all your results.
Briefly, you need to:
- Install Conda
- Download the
.ymlenvironment file - Create and activate the environment
- Deactivate the environment after running your analyses
You can read more about Conda environments and other important concepts to help you make your research reproducible.
 Download and install Conda and Mamba
Download and install Conda and Mamba
Start by installing Conda. We suggest installing Miniconda3 and NOT Anaconda. Follow the instructions for your operating system below (if you need more detailed installation guidelines, you can also check here.
On Mac OS X

curl -o Miniconda3-latest-MacOSX-x86_64.sh https://repo.anaconda.com/miniconda/Miniconda3-latest-MacOSX-x86_64.sh
sh Miniconda3-latest-MacOSX-x86_64.shFollow the instructions on screen replying yes when necessary. Restart your terminal window to apply modifications. After restarting, you can type the command below to install Mamba:
conda init
conda install -n base -c conda-forge mambaOn Ubuntu
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
sh Miniconda3-latest-Linux-x86_64.shFollow the instructions on screen replying yes when necessary. Restart your terminal window to apply modifications. After restarting, you can type the command below to install Mamba:
conda init
conda install -n base -c conda-forge mambaOn Windows 10
![]()
Unfortunately, not all packages available on conda are compatible with windows machines. The good news is that Windows 10 offers native linux support via the Windows Subsystem for Linux (WSL2). This allows you to run linux/bash commands from within windows without the need of a virtual machine nor a dual-boot setup (i.e. having 2 operating systems). However, WSL does not offer a complete support for graphical interfaces (such as RStudio in our case), so we need additional steps to make that happen.
On Windows 10, install the WSL if you don’t have it. Follow the instructions here: https://docs.microsoft.com/en-us/windows/wsl/install-win10
Once you have that installed, you can download and install MobaXterm (which is the enhanced terminal with graphical capacity): https://mobaxterm.mobatek.net
It is recommended that you INSTALL the program and not use the portable version.Inside MobaXterm, you will probably will see that your WSL is already listed on the left panel as an available connection. Just double-click it and you will be accessing it via MobaXterm. If by any chance you don’t see it there, close MobaXterm and go to the WSL terminal, because probably the WSL is not allowing SSH connections. You can follow this link for the instructions on how to do it. You need to complete until the step
Start or restart the SSH service, while the further steps are optional, but might be useful.Inside MobaXterm, download Conda with the command:
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh- Inside MobaXterm, type the commands below to install Conda. Follow the instructions for the installation there.
sh Miniconda3-latest-Linux-x86_64.sh- Inside MobaXterm, Follow the instructions on screen replying
yeswhen necessary. Restart your terminal window to apply modifications. After restarting, you can type the command below to install Mamba:
conda init
conda install -n base -c conda-forge mamba- Inside MobaXterm, type the commands below to install the X-server graphical packages that will be used to launch RStudio. https://docs.anaconda.com/anaconda/install/linux/
sudo apt-get update
sudo apt-get install libgl1-mesa-glx libegl1-mesa libxrandr2 libxrandr2 libxss1 libxcursor1 libxcomposite1 libasound2 libxi6 libxtst6- Close and open all application and Inside MobaXterm, you will probably will see that your WSL is already listed on the left panel as an available connection. Just double-click it and you will be accessing it via MobaXterm.
VirtualBox
![]()
If by any means you see that the installations are not working as it should on your computer, you can try to create a virtual machine to run UBUNTU and install everything there. But please keep this alternative as the last temporary resourse, as we recommend troubleshooting the installation o the up-mentioned methods.
Download and install on your machine VIRTUALBOX https://www.virtualbox.org
Download the ISO disk of UBUNTU https://ubuntu.com/download/desktop
On VIRTUALBOX, click on
Settings(yellow engine) >General>Advancedand make sure that both settings Shared Clipboard and Drag’n’Drop are set toBidirectional.Completely close VIRTUALBOX and start it again to apply changes.
On VIRTUALBOX, create a machine called Ubuntu and add the image above
- set the memory to the maximum allowed in the GREEN bar
- set the hard disk to be dynamic allocated
- all other things can be default
Proceed with the Ubuntu installation as recommended. You can set to do “Minimal Installation” and deactivate to get updates during installation.
Inside Ubuntu, open TERMINAL and type the commands below to install the X-server graphical packages that will be used to launch RStudio. https://docs.anaconda.com/anaconda/install/linux/
sudo apt-get update
sudo apt-get install libgl1-mesa-glx libegl1-mesa libxrandr2 libxrandr2 libxss1 libxcursor1 libxcomposite1 libasound2 libxi6 libxtst6- Inside UBUNTU, Download conda:
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh- Inside UBUNTU, open the TERMINAL and type the commands below. Follow the instructions for the installation there.
sh Miniconda3-latest-Linux-x86_64.sh- Close Terminal to apply the CONDA updates.
Create a conda environment from file
On Mac OS X
To download the environment definition file using the command on Terminal:
curl -o env_MacOSX.yml https://raw.githubusercontent.com/NBISweden/workshop-RNAseq/master/env_MacOSX.ymlAfter this, you should have a file named env_MacOSX.yml in your directory (it does not matter where). Next, type:
mamba env create -f env_MacOSX.ymlSeveral messages will show up on your screen and will tell you about the installation process. This may take a few minutes depending on how many packages are to be installed.
##Collecting package metadata: done
##Solving environment: done
##
##Downloading and Extracting Packages
##libcblas-3.8.0 | 6 KB | ############################################################################# | 100%
##liblapack-3.8.0 | 6 KB | ############################################################################# | 100%
##...
##Preparing transaction: done
##Verifying transaction: done
##Executing transaction: doneOn Ubuntu
To download the environment definition file using the command on Terminal:
curl -o env_linuxOrMobaXTerm.yml https://raw.githubusercontent.com/NBISweden/workshop-RNAseq/master/env_linuxOrMobaXTerm.ymlAfter this, you should have a file named env_linuxOrMobaXTerm.yml in your directory (it does not matter where). Next, type:
mamba env create -f env_linuxOrMobaXTerm.ymlSeveral messages will show up on your screen and will tell you about the installation process. This may take a few minutes depending on how many packages are to be installed.
##Collecting package metadata: done
##Solving environment: done
##
##Downloading and Extracting Packages
##libcblas-3.8.0 | 6 KB | ############################################################################# | 100%
##liblapack-3.8.0 | 6 KB | ############################################################################# | 100%
##...
##Preparing transaction: done
##Verifying transaction: done
##Executing transaction: doneOn Windows 10
![]()
To download the environment definition file type the command inside MobaXTerm on Terminal:
curl -o env_linuxOrMobaXTerm.yml https://raw.githubusercontent.com/NBISweden/workshop-RNAseq/master/env_linuxOrMobaXTerm.ymlAfter this, you should have a file named env_linuxOrMobaXTerm.yml in your directory (it does not matter where). Next, type:
mamba env create -f env_linuxOrMobaXTerm.ymlSeveral messages will show up on your screen and will tell you about the installation process. This may take a few minutes depending on how many packages are to be installed.
##Collecting package metadata: done
##Solving environment: done
##
##Downloading and Extracting Packages
##libcblas-3.8.0 | 6 KB | ############################################################################# | 100%
##liblapack-3.8.0 | 6 KB | ############################################################################# | 100%
##...
##Preparing transaction: done
##Verifying transaction: done
##Executing transaction: doneVirtualBox
![]()
You can create a course folder, download the environment file and create the environment as follows:
mkdir ~/Desktop/course
cd ~/Desktop/course
wget https://raw.githubusercontent.com/NBISweden/workshop-RNAseq/master/env_linuxOrMobaXTerm.yml
conda env create -f env_linuxOrMobaXTerm.yml Activate the environment
Once the environment is created, we need to activate it in order to use the software and packages inside it. To activate an environment type:
conda activate nbis-workshop-rnaseqFrom this point on you can run any of the contents from the workshop. For instance, you can directly launch RStudio by typing rstudio. Here it is important to add the & symbol in the end to be able to use the command line at the same time if needed. You can open other files from Rstudio later as well.
rstudio PATH/my_script.Rmd & Deactivate the environment
After you’ve ran all your analyses, you can deactivate the environment by typing:
conda deactivateData

The data being used in this workshop is available here. However, we recommend you to download it using terminal, which simplify things a lot.
Go to your home folder and launch the course environment:
cd ~/
conda activate nbis-workshop-rnaseqThen you can simply run the curl command below to download the whole data for the course. This will take a long time since files are quite big.
curl https://export.uppmax.uu.se/uppstore2017171/workshops/RNAseq.2024.tar -o RNAseq.2024.tar
tar -xf RNAseq.2024.tarAfter downloading, you will have a folder Do not rename nor place those folder elsewhere, since they will be used as is. You can now type:
ls ~/RNAseq/Code

The code from the course can be downloaded directly from Github.
But it is much easier to do it from the command line:
mkdir ~/RNAseq ~/RNAseq/labs #Run this line IF you have not downloaded the data for the course yet
cd ~/RNAseq/labs
for i in lab_dge.Rmd lab_download.Rmd lab_eda.Rmd lab_functional.Rmd lab_preprocessing.Rmd lab_geoquery.Rmd lab_kallisto.Rmd
do
curl -o $i https://raw.githubusercontent.com/NBISweden/workshop-RNAseq/master/$i
doneCanvas
Canvas is the learning management system (LMS) that we will use for this course (as well as all other NBIS courses). You will receive an email inviting you to create an account on canvas (if you do not already have one) and to join the course there. Please follow instructions on the email and contact us if you encounter any problem (edu.rnaseq [at] nbis.se).