suppressPackageStartupMessages({
library(Seurat)
library(plotly)
options(rgl.printRglwidget = TRUE)
library(Matrix)
library(sparseMatrixStats)
library(slingshot)
library(tradeSeq)
library(patchwork)
})
# Define some color palette
pal <- c(scales::hue_pal()(8), RColorBrewer::brewer.pal(9, "Set1"), RColorBrewer::brewer.pal(8, "Set2"))
set.seed(1)
pal <- rep(sample(pal, length(pal)), 200)
Note
Code chunks run R commands unless otherwise specified.
1 Loading libraries
Nice function to easily draw a graph:
# Add graph to the base R graphics plot
draw_graph <- function(layout, graph, lwd = 0.2, col = "grey") {
res <- rep(x = 1:(length(graph@p) - 1), times = (graph@p[-1] - graph@p[-length(graph@p)]))
segments(
x0 = layout[graph@i + 1, 1], x1 = layout[res, 1],
y0 = layout[graph@i + 1, 2], y1 = layout[res, 2], lwd = lwd, col = col
)
}2 Preparing data
If you have been using the Seurat, Bioconductor or Scanpy toolkits with your own data, you need to reach to the point where you have:
- A dimensionality reduction on which to run the trajectory (for example: PCA, ICA, MNN, harmony, Diffusion Maps, UMAP)
- The cell clustering information (for example: from Louvain, K-means)
- A KNN/SNN graph (this is useful to inspect and sanity-check your trajectories)
We will be using a subset of a bone marrow dataset (originally containing about 100K cells) for this exercise on trajectory inference.
The bone marrow is the source of adult immune cells, and contains virtually all differentiation stages of cell from the immune system which later circulate in the blood to all other organs.

You can download the data:
# download pre-computed data if missing or long compute
fetch_data <- TRUE
path_trajectory <- "./data/trajectory"
if (!dir.exists(path_trajectory)) dir.create(path_trajectory, recursive = T)
# url for source and intermediate data
path_data <- "https://nextcloud.dc.scilifelab.se/public.php/webdav"
curl_upass <- "-u zbC5fr2LbEZ9rSE:scRNAseq2025"
path_file <- "data/trajectory/trajectory_seurat_filtered.rds"
if (!dir.exists(dirname(path_file))) dir.create(dirname(path_file), recursive = TRUE)
if (!file.exists(path_file)) download.file(url = file.path(path_data, "trajectory/trajectory_seurat_filtered.rds"), destfile = path_file, method = "curl", extra = curl_upass)We already have pre-computed and subsetted the dataset (with 6688 cells and 3585 genes) following the analysis steps in this course. We then saved the objects, so you can use common tools to open and start to work with them (either in R or Python).
In addition there was some manual filtering done to remove clusters that are disconnected and cells that are hard to cluster, which can be seen in this script
3 Reading data
obj <- readRDS("data/trajectory/trajectory_seurat_filtered.rds")
# Calculate cluster centroids (for plotting the labels later)
mm <- sparse.model.matrix(~ 0 + factor(obj$clusters_use))
colnames(mm) <- levels(factor(obj$clusters_use))
centroids2d <- as.matrix(t(t(obj@reductions$umap@cell.embeddings) %*% mm) / Matrix::colSums(mm))Let’s visualize which clusters we have in our dataset:
vars <- c("batches", "dataset", "clusters_use", "Phase")
pl <- list()
for (i in vars) {
pl[[i]] <- DimPlot(obj, group.by = i, label = T) + theme_void() + NoLegend()
}
wrap_plots(pl)
You can check, for example, the number of cells in each cluster:
table(obj$clusters_use)
1 11 12 13 15 16 17 18 2 20 21 25 26 27 28 29 32 33 34 35
128 130 132 168 299 140 256 435 71 98 149 56 154 98 76 125 150 295 147 150
36 38 43 44 46 47 49 5 53 54 55 58 59 6 60 7 8 9
135 128 260 110 140 113 217 100 129 57 247 127 101 160 120 147 120 160 4 Exploring the data
It is crucial that you have some understanding of the dataset being analyzed. What are the clusters you see in your data and most importantly How are the clusters related to each other?. Well, let’s explore the data a bit. With the help of this table, write down which cluster numbers in your dataset express these key markers.
| Marker | Cell Type |
|---|---|
| Cd34 | HSC progenitor |
| Ms4a1 | B cell lineage |
| Cd3e | T cell lineage |
| Ltf | Granulocyte lineage |
| Cst3 | Monocyte lineage |
| Cpa3 | Mast Cell lineage |
| Alas2 | RBC lineage |
| Siglech | Dendritic cell lineage |
| C1qc | Macrophage cell lineage |
| Pf4 | Megakaryocyte cell lineage |
vars <- c("Cd34", "Ms4a1", "Cd3e", "Ltf", "Cst3", "Cpa3", "Alas2", "Siglech", "C1qc", "Pf4")
pl <- list()
pl <- list(DimPlot(obj, group.by = "clusters_use", label = T) + theme_void() + NoLegend())
for (i in vars) {
pl[[i]] <- FeaturePlot(obj, features = i, order = T) + theme_void() + NoLegend()
}
wrap_plots(pl)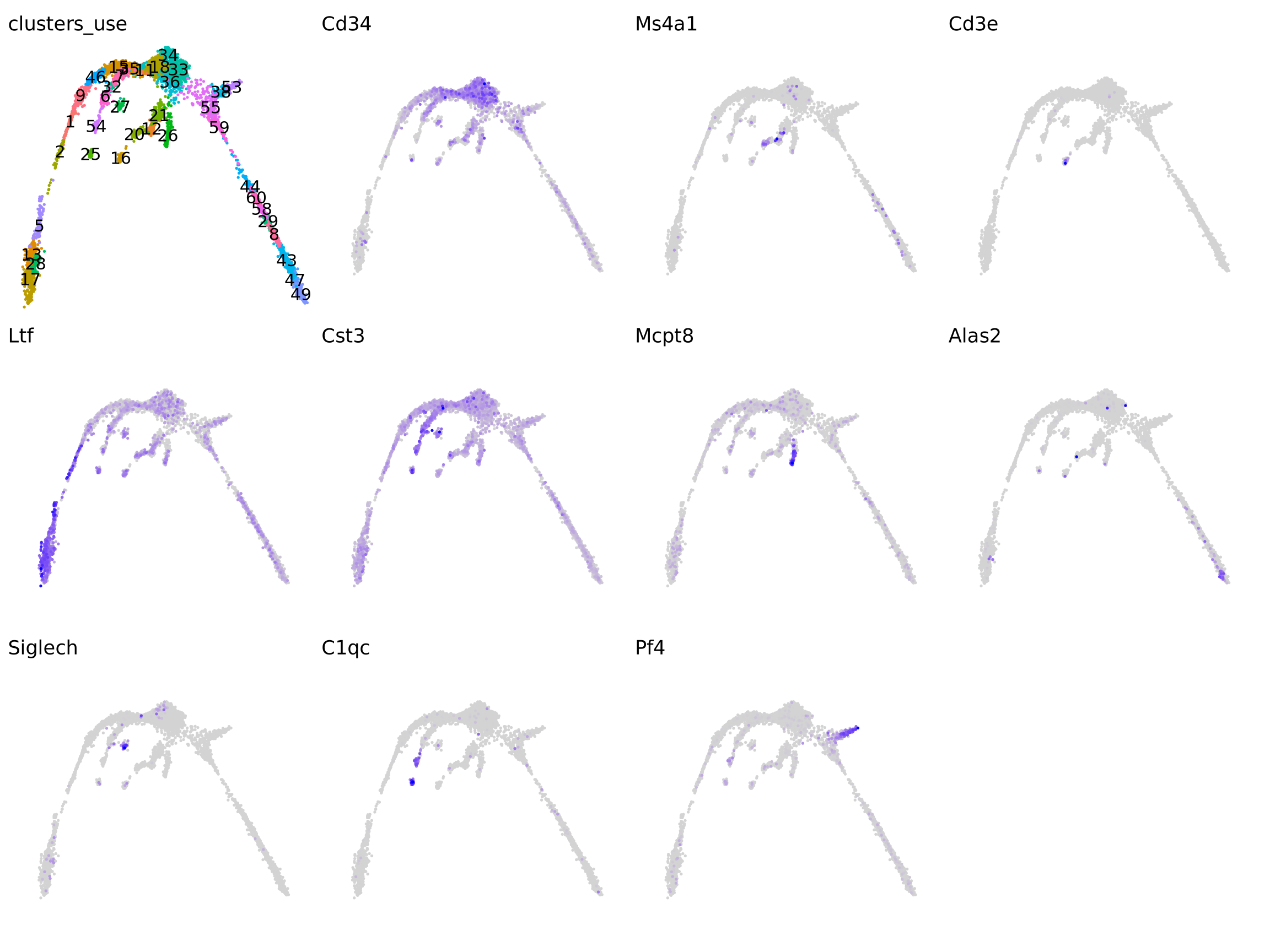
To make it easier to interpret the data, we will add in some labels to the most important clusters.
new_clust = as.character(obj$clusters_use)
new_clust[new_clust == "34"] = "34-Prog" # progenitors
new_clust[new_clust == "17"] = "17-Gran" # granulocytes
new_clust[new_clust == "27"] = "27-DC" # dendritic cells
new_clust[new_clust == "25"] = "25-Mac" # macrophage
new_clust[new_clust == "16"] = "16-TC" # T-cells
new_clust[new_clust == "20"] = "20-BC" # B-cells
new_clust[new_clust == "26"] = "26-MastBaso" # Mast cells / Basophils
new_clust[new_clust == "53"] = "53-Mega" # Megakaryocytes
new_clust[new_clust == "49"] = "49-RBC" # Red blood cells
obj$clust_annot = factor(new_clust)
DimPlot(obj, group.by = "clust_annot", label = T) + theme_void() + NoLegend()
Another way to better explore your data is to look in higher dimensions, to really get a sense for what is right or wrong. As mentioned in the dimensionality reduction exercises, here we ran UMAP with 3 dimensions.
Important
The UMAP needs to be computed to results in exactly 3 dimensions
Since the steps below are identical to both Seurat and Bioconductor toolkits, we will extract the matrices from both, so it is clear what is being used where and to remove long lines of code used to get those matrices. We will use them all. Plot in 3D with Plotly:
df <- data.frame(obj@reductions$umap3d@cell.embeddings, variable = factor(obj$clust_annot))
colnames(df)[1:3] <- c("UMAP_1", "UMAP_2", "UMAP_3")
p_State <- plot_ly(df, x = ~UMAP_1, y = ~UMAP_2, z = ~UMAP_3, color = ~variable, colors = pal, size = .5) %>% add_markers()
p_State# to save interactive plot and open in a new tab
try(htmlwidgets::saveWidget(p_State, selfcontained = T, "data/trajectory/umap_3d_clustering_plotly.html"), silent = T)
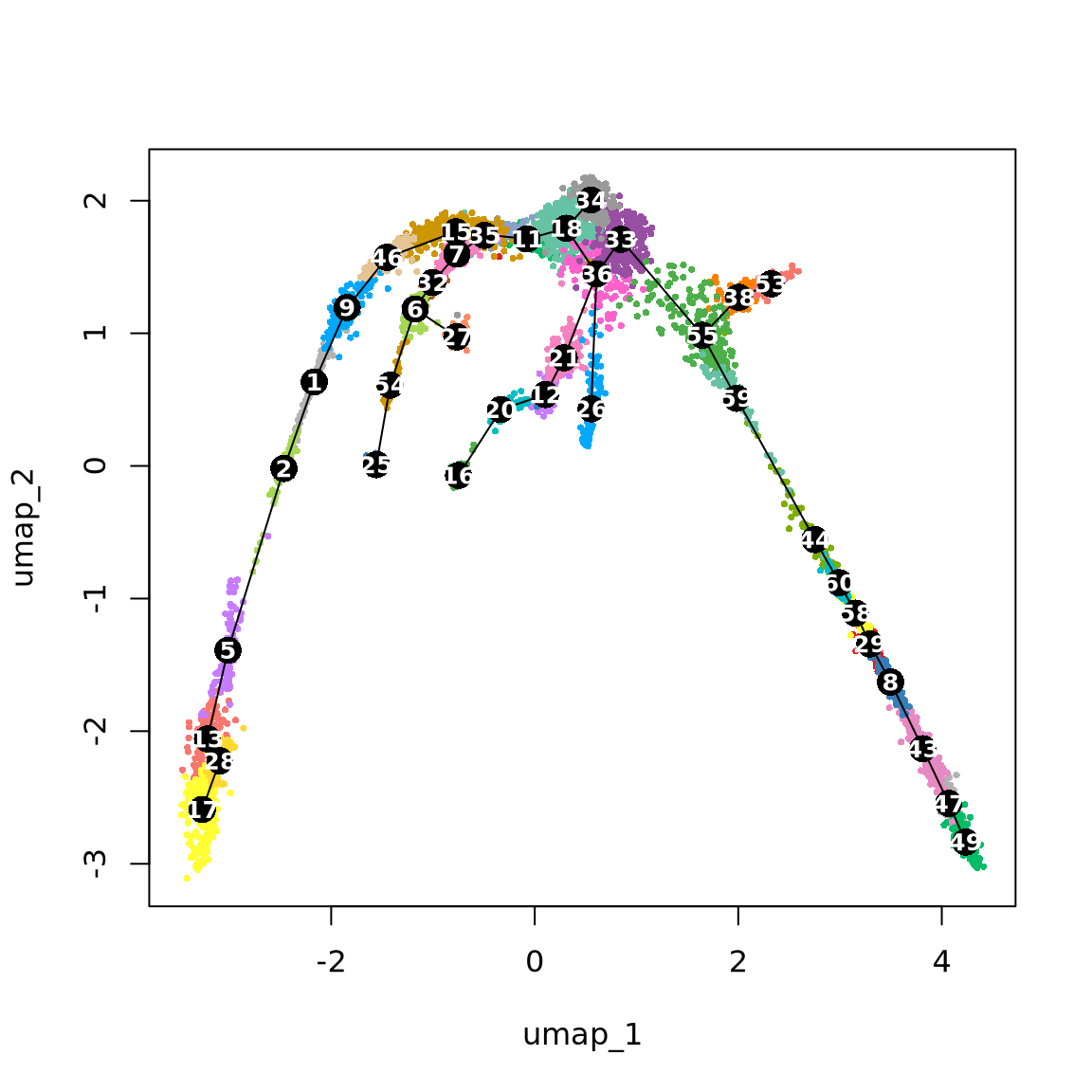
utils::browseURL("data/trajectory/umap_3d_clustering_plotly.html")We can now compute the lineages on these dataset.
# Define lineage ends
ENDS <- c("17-Gran", "27-DC", "25-Mac", "16-TC", "26-MastBaso", "53-Mega", "49-RBC")
set.seed(1)
lineages <- as.SlingshotDataSet(getLineages(
data = obj@reductions$umap3d@cell.embeddings,
clusterLabels = obj$clust_annot,
dist.method = "mnn", # It can be: "simple", "scaled.full", "scaled.diag", "slingshot" or "mnn"
end.clus = ENDS, # You can also define the ENDS!
start.clus = "34-Prog"
)) # define where to START the trajectories
# IF NEEDED, ONE CAN ALSO MANULALLY EDIT THE LINEAGES, FOR EXAMPLE:
# sel <- sapply( lineages@lineages, function(x){rev(x)[1]} ) %in% ENDS
# lineages@lineages <- lineages@lineages[ sel ]
# names(lineages@lineages) <- paste0("Lineage",1:length(lineages@lineages))
# lineages
# Change the reduction to our "fixed" UMAP2d (FOR VISUALISATION ONLY)
lineages@reducedDim <- obj@reductions$umap@cell.embeddings
{
plot(obj@reductions$umap@cell.embeddings, col = pal[obj$clust_annot], cex = .5, pch = 16)
lines(lineages, lwd = 1, col = "black", cex = 2)
text(centroids2d, labels = rownames(centroids2d), cex = 0.8, font = 2, col = "white")
}
5 Defining Principal Curves
Once the clusters are connected, Slingshot allows you to transform them to a smooth trajectory using principal curves. This is an algorithm that iteratively changes an initial curve to better match the data points. It was developed for linear data. To apply it to single-cell data, slingshot adds two enhancements:
- It will run principal curves for each ‘lineage’, which is a set of clusters that go from a defined start cluster to some end cluster
- Lineages with a same set of clusters will be constrained so that their principal curves remain bundled around the overlapping clusters
Since the function getCurves() takes some time to run, we can speed up the convergence of the curve fitting process by reducing the amount of cells to use in each lineage. Ideally you could all cells, but here we had set approx_points to 300 to speed up. Feel free to adjust that for your dataset.
# Define curves
curves <- as.SlingshotDataSet(getCurves(
data = lineages,
thresh = 1e-1,
stretch = 1e-1,
allow.breaks = F,
approx_points = 100
))
curvesclass: SlingshotDataSet
Samples Dimensions
5828 2
lineages: 7
Lineage1: 34-Prog 18 36 33 55 59 44 60 58 29 8 43 47 49-RBC
Lineage2: 34-Prog 18 11 15 46 9 1 2 5 13 28 17-Gran
Lineage3: 34-Prog 18 11 15 35 7 32 6 54 25-Mac
Lineage4: 34-Prog 18 11 15 35 7 32 6 27-DC
Lineage5: 34-Prog 18 36 21 12 20-BC 16-TC
Lineage6: 34-Prog 18 36 33 55 38 53-Mega
Lineage7: 34-Prog 18 36 26-MastBaso
curves: 7
Curve1: Length: 6.7241 Samples: 2161.05
Curve2: Length: 7.3487 Samples: 2097.02
Curve3: Length: 3.5349 Samples: 1502.25
Curve4: Length: 2.5623 Samples: 1387.66
Curve5: Length: 2.9268 Samples: 979.78
Curve6: Length: 2.8976 Samples: 1086.34
Curve7: Length: 2.1323 Samples: 644.86# Plots
{
plot(obj@reductions$umap@cell.embeddings, col = pal[obj$clust_annot], pch = 16)
lines(curves, lwd = 2, col = "black")
text(centroids2d, labels = levels(obj$clust_annot), cex = 1, font = 2)
}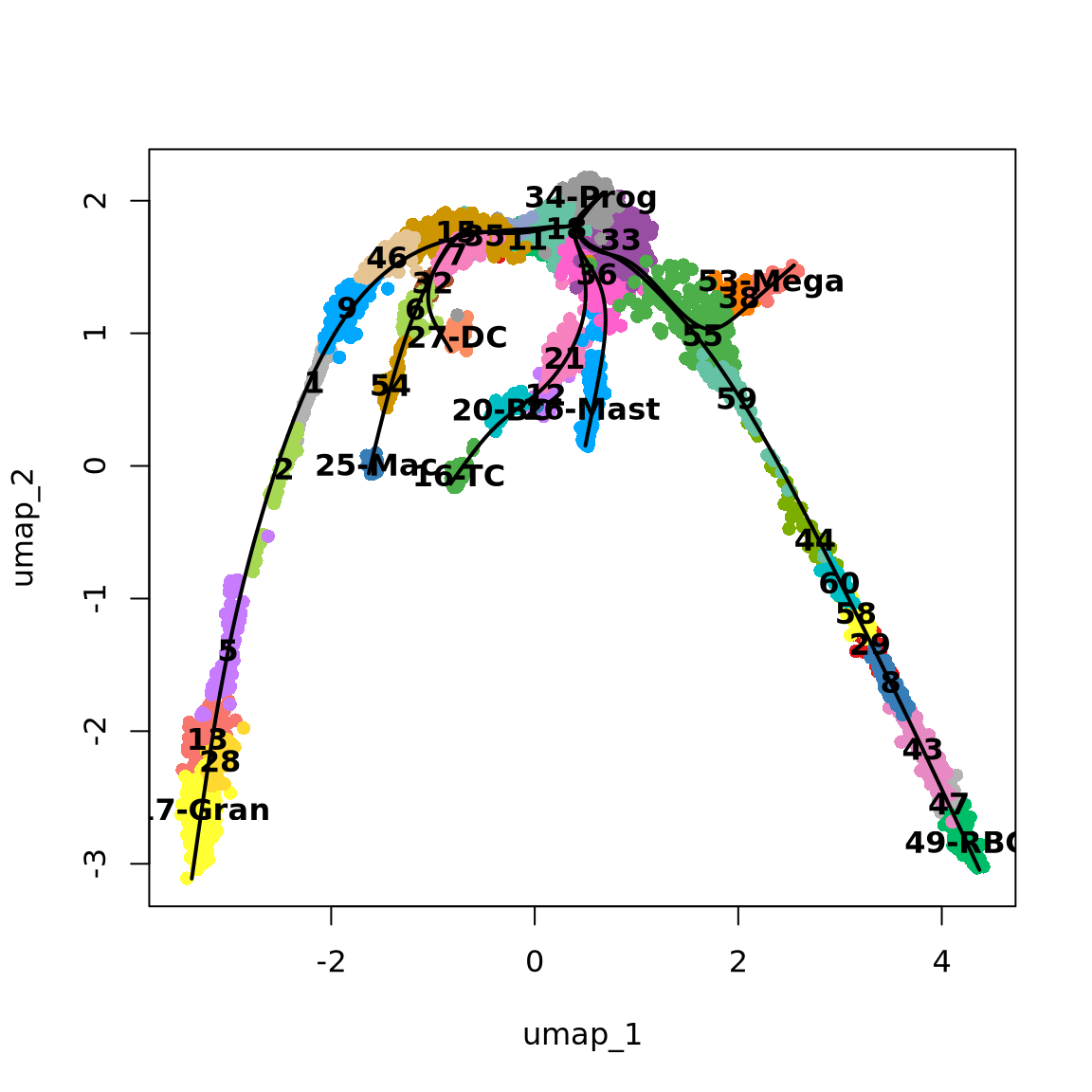
Discuss
Does these lineages fit the biological expectations given what you know of hematopoesis. Please have a look at the figure in Section 2 and compare to the paths you now have.
With those results in hands, we can now compute the differentiation pseudotime.
pseudotime <- slingPseudotime(curves, na = FALSE)
cellWeights <- slingCurveWeights(curves)
x <- rowMeans(pseudotime)
x <- x / max(x)
o <- order(x)
{
plot(obj@reductions$umap@cell.embeddings[o, ],
main = paste0("pseudotime"), pch = 16, cex = 0.4, axes = F, xlab = "", ylab = "",
col = colorRampPalette(c("grey70", "orange3", "firebrick", "purple4"))(99)[x[o] * 98 + 1]
)
points(centroids2d, cex = 2.5, pch = 16, col = "#FFFFFF99")
text(centroids2d, labels = levels(obj$clust_annot), cex = 1, font = 2)
}
Discuss
The pseudotime represents the distance of every cell to the starting cluster. Have a look at the pseudotime plot, how well do you think it represents actual developmental time? What does it represent?
6 Finding differentially expressed genes
The main way to interpret a trajectory is to find genes that change along the trajectory. There are many ways to define differential expression along a trajectory:
- Expression changes along a particular path (i.e. change with pseudotime)
- Expression differences between branches
- Expression changes at branch points
- Expression changes somewhere along the trajectory
- …
tradeSeq is a recently proposed algorithm to find trajectory differentially expressed genes. It works by smoothing the gene expression along the trajectory by fitting a smoother using generalized additive models (GAMs), and testing whether certain coefficients are statistically different between points in the trajectory.
BiocParallel::register(BiocParallel::MulticoreParam())The fitting of GAMs can take quite a while, so for demonstration purposes we first do a very stringent filtering of the genes.
Tip
In an ideal experiment, you would use all the genes, or at least those defined as being variable.
sel_cells <- split(colnames(obj@assays$RNA@data), obj$clust_annot)
sel_cells <- unlist(lapply(sel_cells, function(x) {
set.seed(1)
return(sample(x, 20))
}))
gv <- as.data.frame(na.omit(scran::modelGeneVar(obj@assays$RNA@data[, sel_cells])))
gv <- gv[order(gv$bio, decreasing = T), ]
sel_genes <- sort(rownames(gv)[1:500])Fitting the model:
Caution
This is a slow compute intensive step, we will not run this now and instead use a pre-computed file in the step below.
path_file <- "data/trajectory/seurat_scegam.rds"
# fetch_data is defined at the top of this document
if (!fetch_data) {
sceGAM <- fitGAM(
counts = drop0(obj@assays$RNA@data[sel_genes, sel_cells]),
pseudotime = pseudotime[sel_cells, ],
cellWeights = cellWeights[sel_cells, ],
nknots = 5, verbose = T, parallel = T, sce = TRUE,
BPPARAM = BiocParallel::MulticoreParam()
)
saveRDS(sceGAM, path_file)
}Download the precomputed file.
path_file <- "data/trajectory/seurat_scegam.rds"
# fetch_data is defined at the top of this document
if (fetch_data) {
if (!file.exists(path_file)) download.file(url = file.path(path_data, "trajectory/seurat_scegam.rds"), destfile = path_file, method = "curl", extra = curl_upass)
}# read data
sceGAM <- readRDS(path_file)plotGeneCount(curves, clusters = obj$clust_annot, models = sceGAM)
lineagesclass: SlingshotDataSet
Samples Dimensions
5828 2
lineages: 7
Lineage1: 34-Prog 18 36 33 55 59 44 60 58 29 8 43 47 49-RBC
Lineage2: 34-Prog 18 11 15 46 9 1 2 5 13 28 17-Gran
Lineage3: 34-Prog 18 11 15 35 7 32 6 54 25-Mac
Lineage4: 34-Prog 18 11 15 35 7 32 6 27-DC
Lineage5: 34-Prog 18 36 21 12 20-BC 16-TC
Lineage6: 34-Prog 18 36 33 55 38 53-Mega
Lineage7: 34-Prog 18 36 26-MastBaso
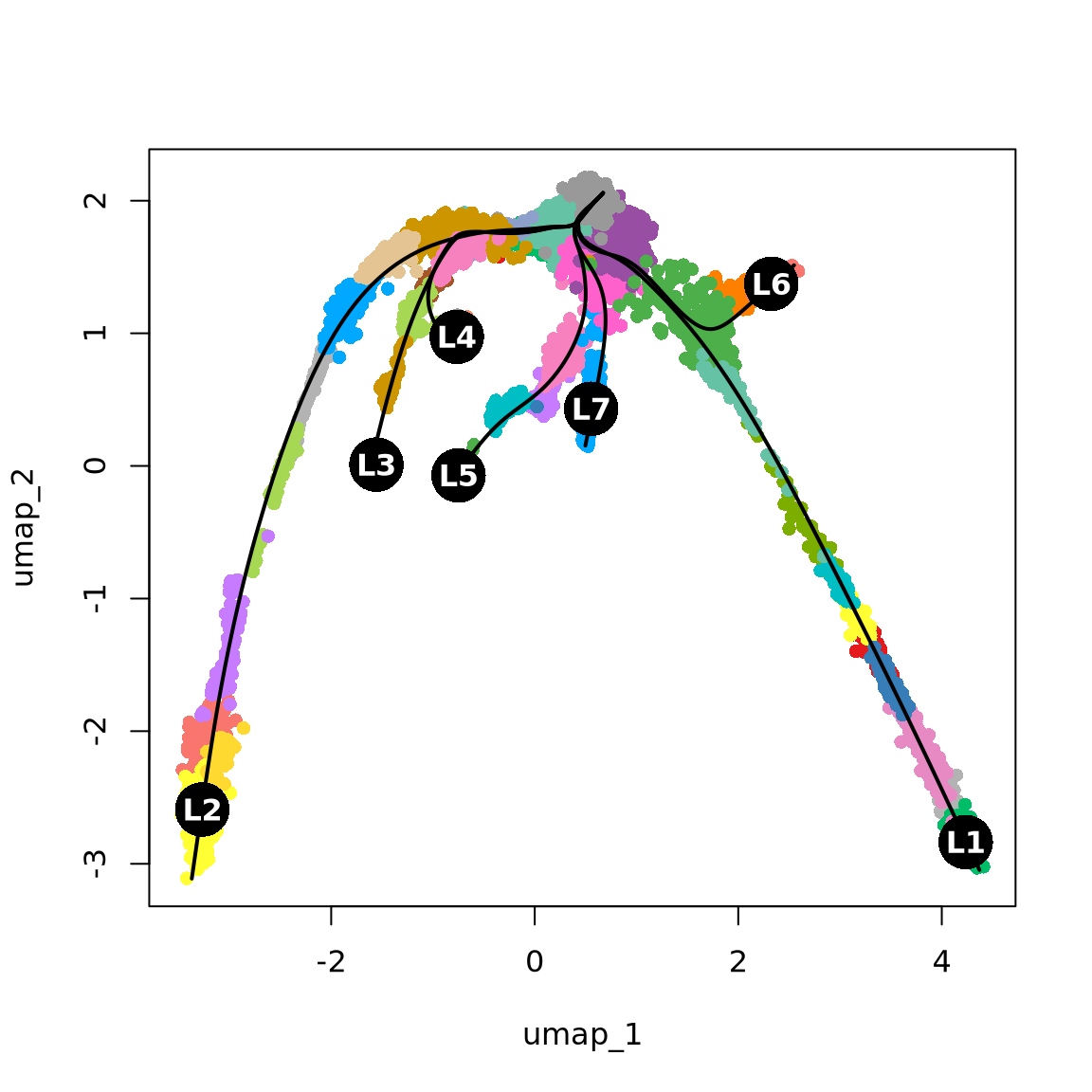
curves: 0 lc <- sapply(lineages@lineages, function(x) {
rev(x)[1]
})
names(lc) <- gsub("Lineage", "L", names(lc))
lc.idx = match(lc, levels(obj$clust_annot))
{
plot(obj@reductions$umap@cell.embeddings, col = pal[obj$clust_annot], pch = 16)
lines(curves, lwd = 2, col = "black")
points(centroids2d[lc.idx, ], col = "black", pch = 16, cex = 4)
text(centroids2d[lc.idx, ], labels = names(lc), cex = 1, font = 2, col = "white")
}
6.1 Genes that change with pseudotime
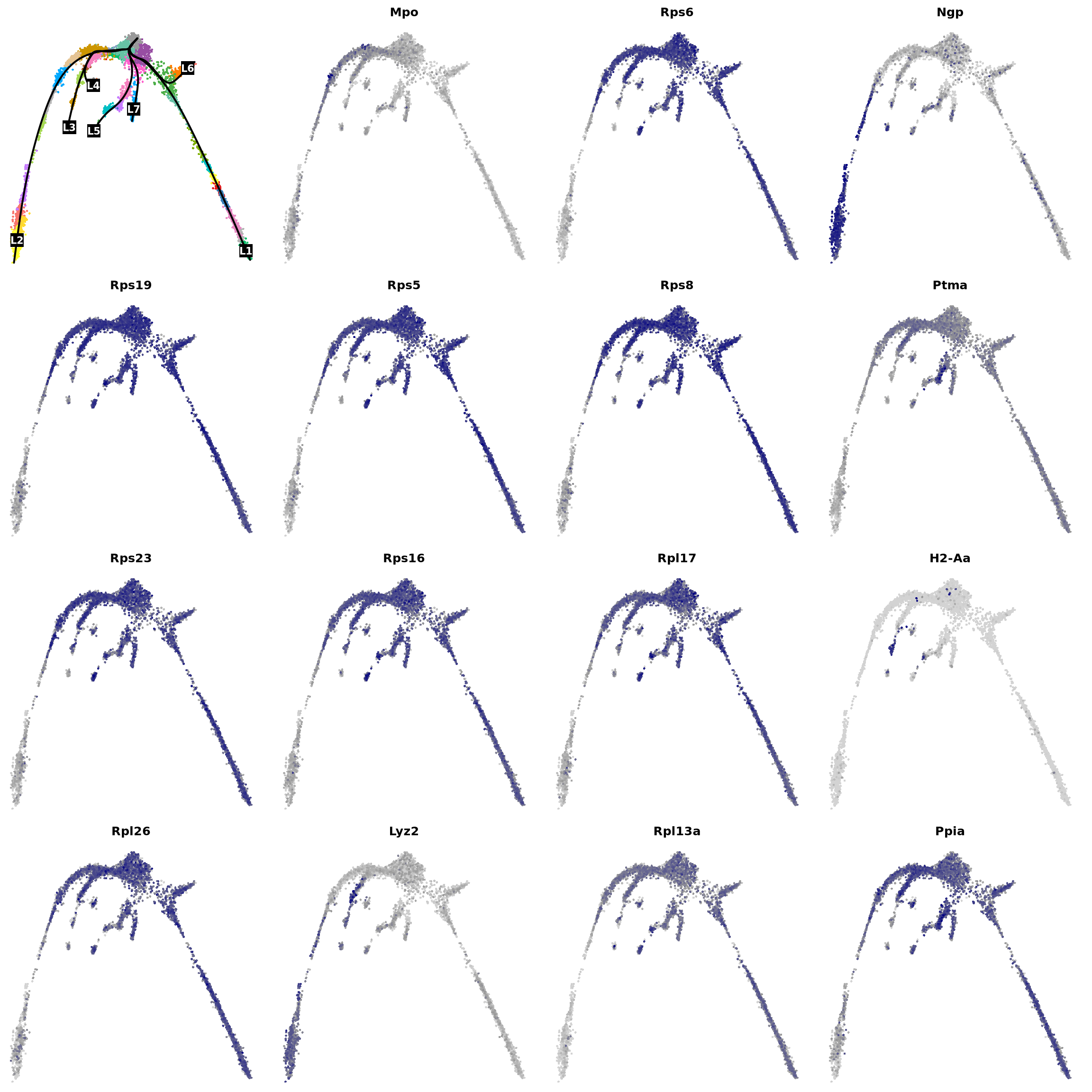
We can first look at general trends of gene expression across pseudotime.
set.seed(8)
res <- na.omit(associationTest(sceGAM, contrastType = "consecutive"))
res <- res[res$pvalue < 1e-3, ]
res <- res[res$waldStat > mean(res$waldStat), ]
res <- res[order(res$waldStat, decreasing = T), ]
res[1:10, ]We can plot their expression:
par(mfrow = c(4, 4), mar = c(.1, .1, 2, 1))
{
plot(obj@reductions$umap@cell.embeddings, col = pal[obj$clusters_use], cex = .5, pch = 16, axes = F, xlab = "", ylab = "")
lines(curves, lwd = 2, col = "black")
points(centroids2d[lc.idx, ], col = "black", pch = 15, cex = 3, xpd = T)
text(centroids2d[lc.idx, ], labels = names(lc), cex = 1, font = 2, col = "white", xpd = T)
}
vars <- rownames(res[1:15, ])
vars <- na.omit(vars[vars != "NA"])
for (i in vars) {
x <- drop0(obj@assays$RNA@data)[i, ]
x <- (x - min(x)) / (max(x) - min(x))
o <- order(x)
plot(obj@reductions$umap@cell.embeddings[o, ],
main = paste0(i), pch = 16, cex = 0.5, axes = F, xlab = "", ylab = "",
col = colorRampPalette(c("lightgray", "grey60", "navy"))(99)[x[o] * 98 + 1]
)
}
6.2 Genes that change between two pseudotime points
We can define custom pseudotime values of interest if we’re interested in genes that change between particular point in pseudotime. By default, we can look at differences between start and end:
res <- na.omit(startVsEndTest(sceGAM, pseudotimeValues = c(0, 1)))
res <- res[res$pvalue < 1e-3, ]
res <- res[res$waldStat > mean(res$waldStat), ]
res <- res[order(res$waldStat, decreasing = T), ]
res[1:10, 1:6]You can see now that there are several more columns, one for each lineage. This table represents the differential expression within each lineage, to identify which genes go up or down. Let’s check lineage 1:
# Get the top UP and Down regulated in lineage 1
res_lin1 <- sort(setNames(res$logFClineage1, rownames(res)))
vars <- names(c(rev(res_lin1)[1:7], res_lin1[1:8]))
vars <- na.omit(vars[vars != "NA"])
par(mfrow = c(4, 4), mar = c(.1, .1, 2, 1))
{
plot(obj@reductions$umap@cell.embeddings, col = pal[obj$clusters_use], cex = .5, pch = 16, axes = F, xlab = "", ylab = "")
lines(curves, lwd = 2, col = "black")
points(centroids2d[lc.idx, ], col = "black", pch = 15, cex = 3, xpd = T)
text(centroids2d[lc.idx, ], labels = names(lc), cex = 1, font = 2, col = "white", xpd = T)
}
for (i in vars) {
x <- drop0(obj@assays$RNA@data)[i, ]
x <- (x - min(x)) / (max(x) - min(x))
o <- order(x)
plot(obj@reductions$umap@cell.embeddings[o, ],
main = paste0(i), pch = 16, cex = 0.5, axes = F, xlab = "", ylab = "",
col = colorRampPalette(c("lightgray", "grey60", "navy"))(99)[x[o] * 98 + 1]
)
}
6.3 Genes that are different between lineages
More interesting are genes that are different between two branches. We may have seen some of these genes already pop up in previous analyses of pseudotime. There are several ways to define “different between branches”, and each have their own functions:
- Different at the end points, using
diffEndTest() - Different at the branching point, using
earlyDETest() - Different somewhere in pseudotime the branching point, using
patternTest()
Note that the last function requires that the pseudotimes between two lineages are aligned.
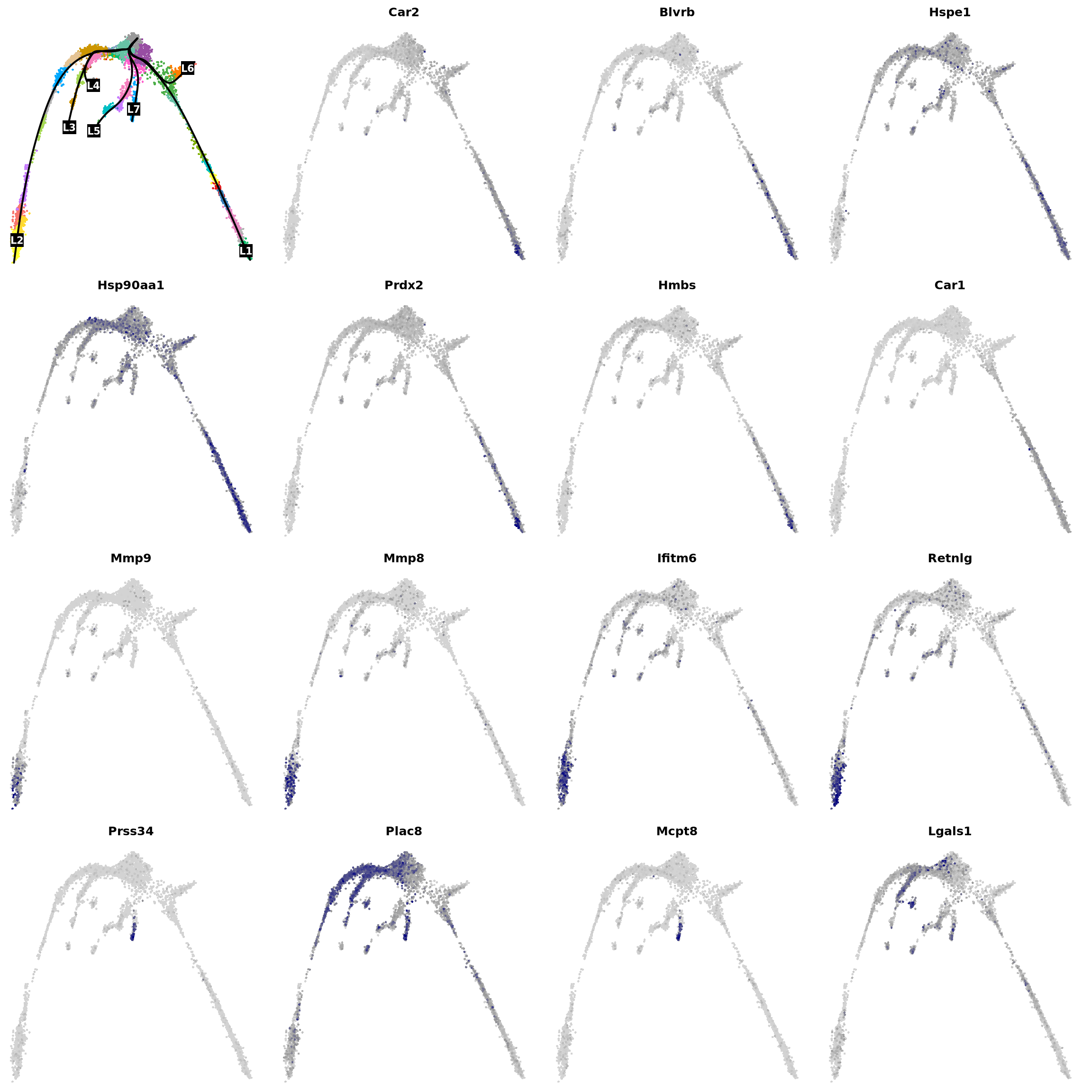
res <- na.omit(diffEndTest(sceGAM))
res <- res[res$pvalue < 1e-3, ]
res <- res[res$waldStat > mean(res$waldStat), ]
res <- res[order(res$waldStat, decreasing = T), ]
res[1:10, ]You can see now that there are even more columns, one for the pairwise comparison between each lineage. Let’s check lineage 1 vs lineage 2:
# Get the top UP and Down regulated in lineage 1 vs 2
res_lin1_2 <- sort(setNames(res$logFC1_2, rownames(res)))
vars <- names(c(rev(res_lin1_2)[1:7], res_lin1_2[1:8]))
vars <- na.omit(vars[vars != "NA"])
par(mfrow = c(4, 4), mar = c(.1, .1, 2, 1))
{
plot(obj@reductions$umap@cell.embeddings, col = pal[obj$clusters_use], cex = .5, pch = 16, axes = F, xlab = "", ylab = "")
lines(curves, lwd = 2, col = "black")
points(centroids2d[lc.idx, ], col = "black", pch = 15, cex = 3, xpd = T)
text(centroids2d[lc.idx, ], labels = names(lc), cex = 1, font = 2, col = "white", xpd = T)
}
for (i in vars) {
x <- drop0(obj@assays$RNA@data)[i, ]
x <- (x - min(x)) / (max(x) - min(x))
o <- order(x)
plot(obj@reductions$umap@cell.embeddings[o, ],
main = paste0(i), pch = 16, cex = 0.5, axes = F, xlab = "", ylab = "",
col = colorRampPalette(c("lightgray", "grey60", "navy"))(99)[x[o] * 98 + 1]
)
}
Check out this vignette for a more in-depth overview of tradeSeq and many other differential expression tests.
7 Generating batch-corrected data for differential gene expression
Before computing differential gene expression, sometimes it is a good idea to make sure our dataset is somewhat homogeneous (without very strong batch effects). In this dataset, we actually used data from 4 different technologies (Drop-seq, SmartSeq2 and 10X) and therefore massive differences in read counts can be observed:
If you want to know more about how to control for this issue, please have a look at batch_corrected_counts.Rmd
8 References
Cannoodt, Robrecht, Wouter Saelens, and Yvan Saeys. 2016. “Computational Methods for Trajectory Inference from Single-Cell Transcriptomics.” European Journal of Immunology 46 (11): 2496–2506. doi.
Saelens, Wouter, Robrecht Cannoodt, Helena Todorov, and Yvan Saeys. 2019. “A Comparison of Single-Cell Trajectory Inference Methods.” Nature Biotechnology 37 (5): 547–54. doi.
9 Session info
Click here
sessionInfo()R version 4.3.3 (2024-02-29)
Platform: x86_64-conda-linux-gnu (64-bit)
Running under: Ubuntu 20.04.6 LTS
Matrix products: default
BLAS/LAPACK: /usr/local/conda/envs/seurat/lib/libopenblasp-r0.3.28.so; LAPACK version 3.12.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] patchwork_1.3.0 tradeSeq_1.16.0
[3] slingshot_2.10.0 TrajectoryUtils_1.10.0
[5] SingleCellExperiment_1.24.0 SummarizedExperiment_1.32.0
[7] Biobase_2.62.0 GenomicRanges_1.54.1
[9] GenomeInfoDb_1.38.1 IRanges_2.36.0
[11] S4Vectors_0.40.2 BiocGenerics_0.48.1
[13] princurve_2.1.6 sparseMatrixStats_1.14.0
[15] MatrixGenerics_1.14.0 matrixStats_1.5.0
[17] Matrix_1.6-5 plotly_4.10.4
[19] ggplot2_3.5.1 Seurat_5.1.0
[21] SeuratObject_5.0.2 sp_2.2-0
loaded via a namespace (and not attached):
[1] RcppAnnoy_0.0.22 splines_4.3.3
[3] later_1.4.1 bitops_1.0-9
[5] tibble_3.2.1 polyclip_1.10-7
[7] fastDummies_1.7.5 lifecycle_1.0.4
[9] edgeR_4.0.16 globals_0.16.3
[11] lattice_0.22-6 MASS_7.3-60.0.1
[13] crosstalk_1.2.1 magrittr_2.0.3
[15] limma_3.58.1 rmarkdown_2.29
[17] yaml_2.3.10 metapod_1.10.0
[19] httpuv_1.6.15 sctransform_0.4.1
[21] spam_2.11-1 spatstat.sparse_3.1-0
[23] reticulate_1.40.0 cowplot_1.1.3
[25] pbapply_1.7-2 RColorBrewer_1.1-3
[27] abind_1.4-5 zlibbioc_1.48.0
[29] Rtsne_0.17 purrr_1.0.2
[31] RCurl_1.98-1.16 GenomeInfoDbData_1.2.11
[33] ggrepel_0.9.6 irlba_2.3.5.1
[35] listenv_0.9.1 spatstat.utils_3.1-2
[37] goftest_1.2-3 RSpectra_0.16-2
[39] spatstat.random_3.3-2 dqrng_0.3.2
[41] fitdistrplus_1.2-2 parallelly_1.42.0
[43] DelayedMatrixStats_1.24.0 leiden_0.4.3.1
[45] codetools_0.2-20 DelayedArray_0.28.0
[47] scuttle_1.12.0 tidyselect_1.2.1
[49] farver_2.1.2 ScaledMatrix_1.10.0
[51] viridis_0.6.5 spatstat.explore_3.3-4
[53] jsonlite_1.8.9 BiocNeighbors_1.20.0
[55] progressr_0.15.1 ggridges_0.5.6
[57] survival_3.8-3 tools_4.3.3
[59] ica_1.0-3 Rcpp_1.0.14
[61] glue_1.8.0 gridExtra_2.3
[63] SparseArray_1.2.2 xfun_0.50
[65] mgcv_1.9-1 dplyr_1.1.4
[67] withr_3.0.2 fastmap_1.2.0
[69] bluster_1.12.0 digest_0.6.37
[71] rsvd_1.0.5 R6_2.6.1
[73] mime_0.12 colorspace_2.1-1
[75] scattermore_1.2 tensor_1.5
[77] spatstat.data_3.1-4 tidyr_1.3.1
[79] generics_0.1.3 data.table_1.16.4
[81] httr_1.4.7 htmlwidgets_1.6.4
[83] S4Arrays_1.2.0 uwot_0.2.2
[85] pkgconfig_2.0.3 gtable_0.3.6
[87] lmtest_0.9-40 XVector_0.42.0
[89] htmltools_0.5.8.1 dotCall64_1.2
[91] scales_1.3.0 png_0.1-8
[93] spatstat.univar_3.1-1 scran_1.30.0
[95] knitr_1.49 reshape2_1.4.4
[97] nlme_3.1-167 zoo_1.8-12
[99] stringr_1.5.1 KernSmooth_2.23-26
[101] parallel_4.3.3 miniUI_0.1.1.1
[103] pillar_1.10.1 grid_4.3.3
[105] vctrs_0.6.5 RANN_2.6.2
[107] promises_1.3.2 BiocSingular_1.18.0
[109] beachmat_2.18.0 xtable_1.8-4
[111] cluster_2.1.8 evaluate_1.0.3
[113] cli_3.6.4 locfit_1.5-9.11
[115] compiler_4.3.3 rlang_1.1.5
[117] crayon_1.5.3 future.apply_1.11.3
[119] labeling_0.4.3 plyr_1.8.9
[121] stringi_1.8.4 viridisLite_0.4.2
[123] deldir_2.0-4 BiocParallel_1.36.0
[125] munsell_0.5.1 lazyeval_0.2.2
[127] spatstat.geom_3.3-5 RcppHNSW_0.6.0
[129] future_1.34.0 statmod_1.5.0
[131] shiny_1.10.0 ROCR_1.0-11
[133] igraph_2.0.3