import os
path_data = "https://nextcloud.dc.scilifelab.se/public.php/webdav"
curl_upass = "zbC5fr2LbEZ9rSE:scRNAseq2025"
path_covid = "./data/covid/raw"
if not os.path.exists(path_covid):
os.makedirs(path_covid, exist_ok=True)
path_results = "data/covid/results"
if not os.path.exists(path_results):
os.makedirs(path_results, exist_ok=True)
Note
Code chunks run Python commands unless it starts with %%bash, in which case, those chunks run shell commands.
1 Get data
In this tutorial, we will run all steps with a set of 8 PBMC 10x datasets from 4 covid-19 patients and 4 healthy controls, the samples have been subsampled to 1500 cells per sample. We can start by defining our paths.
import subprocess
file_list = [
"normal_pbmc_13.h5", "normal_pbmc_14.h5", "normal_pbmc_19.h5", "normal_pbmc_5.h5",
"ncov_pbmc_15.h5", "ncov_pbmc_16.h5", "ncov_pbmc_17.h5", "ncov_pbmc_1.h5"
]
for i in file_list:
path_file = os.path.join(path_covid, i)
if not os.path.exists(path_file):
file_url = os.path.join(path_data, "covid/raw", i)
subprocess.call(["curl", "-u", curl_upass, "-o", path_file, file_url ])With data in place, now we can start loading libraries we will use in this tutorial.
import numpy as np
import pandas as pd
import scanpy as sc
import warnings
import gc
warnings.simplefilter(action='ignore', category=Warning)
# verbosity: errors (0), warnings (1), info (2), hints (3)
sc.settings.verbosity = 3
sc.settings.set_figure_params(dpi=80)We can first load the data individually by reading directly from HDF5 file format (.h5).
In Scanpy we read them into an Anndata object with the the function read_10x_h5
data_cov1 = sc.read_10x_h5(os.path.join(path_covid,'ncov_pbmc_1.h5'))
data_cov1.var_names_make_unique()
data_cov15 = sc.read_10x_h5(os.path.join(path_covid,'ncov_pbmc_15.h5'))
data_cov15.var_names_make_unique()
data_cov16 = sc.read_10x_h5(os.path.join(path_covid,'ncov_pbmc_16.h5'))
data_cov16.var_names_make_unique()
data_cov17 = sc.read_10x_h5(os.path.join(path_covid,'ncov_pbmc_17.h5'))
data_cov17.var_names_make_unique()
data_ctrl5 = sc.read_10x_h5(os.path.join(path_covid,'normal_pbmc_5.h5'))
data_ctrl5.var_names_make_unique()
data_ctrl13 = sc.read_10x_h5(os.path.join(path_covid,'normal_pbmc_13.h5'))
data_ctrl13.var_names_make_unique()
data_ctrl14 = sc.read_10x_h5(os.path.join(path_covid,'normal_pbmc_14.h5'))
data_ctrl14.var_names_make_unique()
data_ctrl19 = sc.read_10x_h5(os.path.join(path_covid,'normal_pbmc_19.h5'))
data_ctrl19.var_names_make_unique()reading data/covid/raw/ncov_pbmc_1.h5
(0:00:00)
reading data/covid/raw/ncov_pbmc_15.h5
(0:00:00)
reading data/covid/raw/ncov_pbmc_16.h5
(0:00:00)
reading data/covid/raw/ncov_pbmc_17.h5
(0:00:00)
reading data/covid/raw/normal_pbmc_5.h5
(0:00:00)
reading data/covid/raw/normal_pbmc_13.h5
(0:00:00)
reading data/covid/raw/normal_pbmc_14.h5
(0:00:00)
reading data/covid/raw/normal_pbmc_19.h5
(0:00:00)2 Collate
We can now merge them objects into a single object. Each analysis workflow (Seurat, Scater, Scanpy, etc) has its own way of storing data. We will add dataset labels as cell.ids just in case you have overlapping barcodes between the datasets. After that we add a column type in the metadata to define covid and ctrl samples.
# add some metadata
data_cov1.obs['type']="Covid"
data_cov1.obs['sample']="covid_1"
data_cov15.obs['type']="Covid"
data_cov15.obs['sample']="covid_15"
data_cov16.obs['type']="Covid"
data_cov16.obs['sample']="covid_16"
data_cov17.obs['type']="Covid"
data_cov17.obs['sample']="covid_17"
data_ctrl5.obs['type']="Ctrl"
data_ctrl5.obs['sample']="ctrl_5"
data_ctrl13.obs['type']="Ctrl"
data_ctrl13.obs['sample']="ctrl_13"
data_ctrl14.obs['type']="Ctrl"
data_ctrl14.obs['sample']="ctrl_14"
data_ctrl19.obs['type']="Ctrl"
data_ctrl19.obs['sample']="ctrl_19"
# merge into one object.
adata = data_cov1.concatenate(data_cov15, data_cov16, data_cov17, data_ctrl5, data_ctrl13, data_ctrl14, data_ctrl19)
# and delete individual datasets to save space
del(data_cov1, data_cov15, data_cov16, data_cov17)
del(data_ctrl5, data_ctrl13, data_ctrl14, data_ctrl19)
gc.collect()413You can print a summary of the datasets in the Scanpy object, or a summary of the whole object.
print(adata.obs['sample'].value_counts())
adatasample
covid_1 1500
covid_15 1500
covid_16 1500
covid_17 1500
ctrl_5 1500
ctrl_13 1500
ctrl_14 1500
ctrl_19 1500
Name: count, dtype: int64AnnData object with n_obs × n_vars = 12000 × 33538
obs: 'type', 'sample', 'batch'
var: 'gene_ids', 'feature_types', 'genome'3 Calculate QC
Having the data in a suitable format, we can start calculating some quality metrics. We can for example calculate the percentage of mitochondrial and ribosomal genes per cell and add to the metadata. The proportion of hemoglobin genes can give an indication of red blood cell contamination, but in some tissues it can also be the case that some celltypes have higher content of hemoglobin. This will be helpful to visualize them across different metadata parameters (i.e. datasetID and chemistry version). There are several ways of doing this. The QC metrics are finally added to the metadata table.
Citing from Simple Single Cell workflows (Lun, McCarthy & Marioni, 2017) about mitochondrial reads: High proportions are indicative of poor-quality cells (Islam et al. 2014; Ilicic et al. 2016), possibly because of loss of cytoplasmic RNA from perforated cells. The reasoning is that mitochondria are larger than individual transcript molecules and less likely to escape through tears in the cell membrane.
First, let Scanpy calculate some general qc-stats for genes and cells with the function sc.pp.calculate_qc_metrics, similar to calculateQCmetrics() in Scater. It can also calculate proportion of counts for specific gene populations, so first we need to define which genes are mitochondrial, ribosomal and hemoglobin.
# mitochondrial genes
adata.var['mt'] = adata.var_names.str.startswith('MT-')
# ribosomal genes
adata.var['ribo'] = adata.var_names.str.startswith(("RPS","RPL"))
# hemoglobin genes.
adata.var['hb'] = adata.var_names.str.contains(("^HB[^(P|E|S)]"))
adata.var| gene_ids | feature_types | genome | mt | ribo | hb | |
|---|---|---|---|---|---|---|
| MIR1302-2HG | ENSG00000243485 | Gene Expression | GRCh38 | False | False | False |
| FAM138A | ENSG00000237613 | Gene Expression | GRCh38 | False | False | False |
| OR4F5 | ENSG00000186092 | Gene Expression | GRCh38 | False | False | False |
| AL627309.1 | ENSG00000238009 | Gene Expression | GRCh38 | False | False | False |
| AL627309.3 | ENSG00000239945 | Gene Expression | GRCh38 | False | False | False |
| ... | ... | ... | ... | ... | ... | ... |
| AC233755.2 | ENSG00000277856 | Gene Expression | GRCh38 | False | False | False |
| AC233755.1 | ENSG00000275063 | Gene Expression | GRCh38 | False | False | False |
| AC240274.1 | ENSG00000271254 | Gene Expression | GRCh38 | False | False | False |
| AC213203.1 | ENSG00000277475 | Gene Expression | GRCh38 | False | False | False |
| FAM231C | ENSG00000268674 | Gene Expression | GRCh38 | False | False | False |
33538 rows × 6 columns
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt','ribo','hb'], percent_top=None, log1p=False, inplace=True)Now you can see that we have additional data in the metadata slot.
Another opition to using the calculate_qc_metrics function is to calculate the values on your own and add to a metadata slot. An example for mito genes can be found below:
mito_genes = adata.var_names.str.startswith('MT-')
# for each cell compute fraction of counts in mito genes vs. all genes
# the `.A1` is only necessary as X is sparse (to transform to a dense array after summing)
adata.obs['percent_mt2'] = np.sum(
adata[:, mito_genes].X, axis=1).A1 / np.sum(adata.X, axis=1).A1
# add the total counts per cell as observations-annotation to adata
adata.obs['n_counts'] = adata.X.sum(axis=1).A1
adataAnnData object with n_obs × n_vars = 12000 × 33538
obs: 'type', 'sample', 'batch', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'pct_counts_hb', 'percent_mt2', 'n_counts'
var: 'gene_ids', 'feature_types', 'genome', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts'4 Plot QC
Now we can plot some of the QC variables as violin plots.
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt', 'pct_counts_ribo', 'pct_counts_hb'], jitter=0.4, groupby = 'sample', rotation= 45)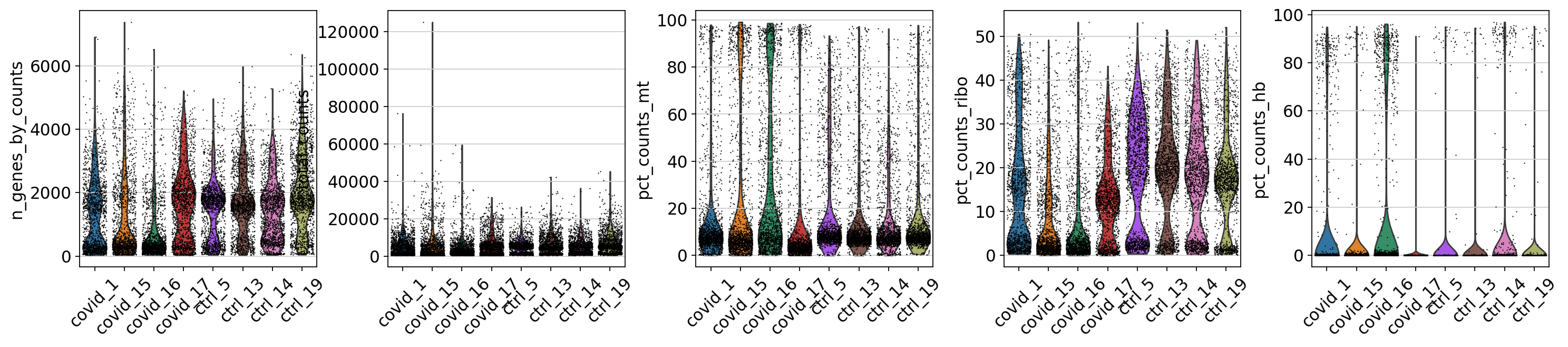
NoteDiscuss
Looking at the violin plots, what do you think are appropriate cutoffs for filtering these samples?
As you can see, there is quite some difference in quality for these samples, with for instance the covid_15 and covid_16 samples having cells with fewer detected genes and more mitochondrial content. We can also plot the different QC-measures as scatter plots.
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt', color="sample")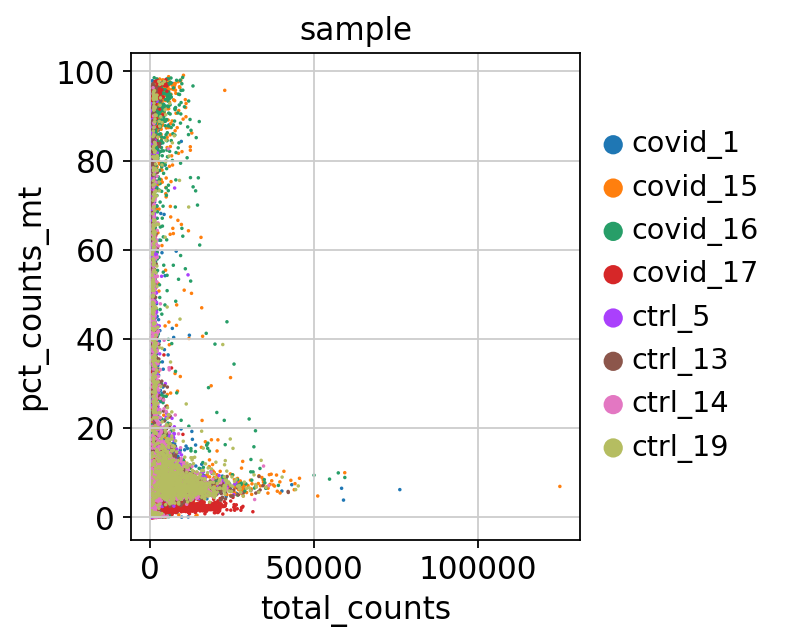
NoteDiscuss
Plot additional QC stats that we have calculated as scatter plots. How are the different measures correlated? Can you explain why?
5 Filtering
5.1 Detection-based filtering
A standard approach is to filter cells with low number of reads as well as genes that are present in at least a given number of cells. Here we will only consider cells with at least 200 detected genes and genes that are expressed in at least 3 cells. Please note that those values are highly dependent on the library preparation method used.
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
print(adata.n_obs, adata.n_vars)filtered out 1336 cells that have less than 200 genes expressed
filtered out 14047 genes that are detected in less than 3 cells
10664 19491Extremely high number of detected genes could indicate doublets. However, depending on the cell type composition in your sample, you may have cells with higher number of genes (and also higher counts) from one cell type. In this case, we will run doublet prediction further down, so we will skip this step now, but the code below is an example of how it can be run:
# skip for now as we are doing doublet prediction
#keep_v2 = (adata.obs['n_genes_by_counts'] < 2000) & (adata.obs['n_genes_by_counts'] > 500) & (adata.obs['lib_prep'] == 'v2')
#print(sum(keep_v2))
# filter for gene detection for v3
#keep_v3 = (adata.obs['n_genes_by_counts'] < 4100) & (adata.obs['n_genes_by_counts'] > 1000) & (adata.obs['lib_prep'] != 'v2')
#print(sum(keep_v3))
# keep both sets of cells
#keep = (keep_v2) | (keep_v3)
#print(sum(keep))
#adata = adata[keep, :]
#print("Remaining cells %d"%adata.n_obs)Additionally, we can also see which genes contribute the most to such reads. We can for instance plot the percentage of counts per gene.
sc.pl.highest_expr_genes(adata, n_top=20)normalizing counts per cell
finished (0:00:02)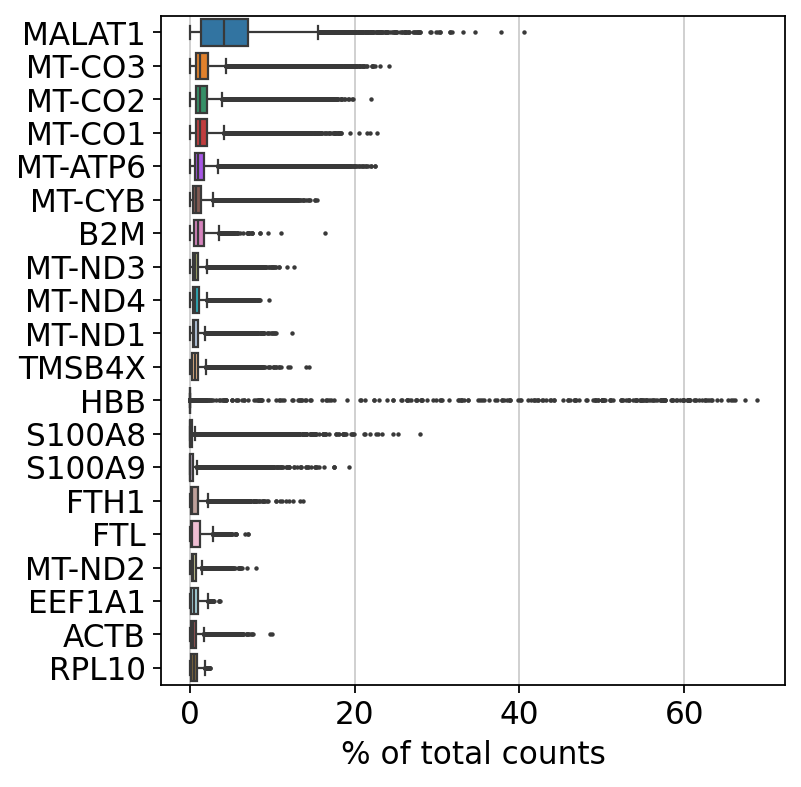
As you can see, MALAT1 constitutes up to 30% of the UMIs from a single cell and the other top genes are mitochondrial and ribosomal genes. It is quite common that nuclear lincRNAs have correlation with quality and mitochondrial reads, so high detection of MALAT1 may be a technical issue. Let us assemble some information about such genes, which are important for quality control and downstream filtering.
5.2 Mito/Ribo filtering
We also have quite a lot of cells with high proportion of mitochondrial and low proportion of ribosomal reads. It would be wise to remove those cells, if we have enough cells left after filtering. Another option would be to either remove all mitochondrial reads from the dataset and hope that the remaining genes still have enough biological signal. A third option would be to just regress out the percent_mito variable during scaling. In this case we had as much as 99.7% mitochondrial reads in some of the cells, so it is quite unlikely that there is much cell type signature left in those. Looking at the plots, make reasonable decisions on where to draw the cutoff. In this case, the bulk of the cells are below 20% mitochondrial reads and that will be used as a cutoff. We will also remove cells with less than 5% ribosomal reads.
# filter for percent mito
adata = adata[adata.obs['pct_counts_mt'] < 20, :]
# filter for percent ribo > 0.05
adata = adata[adata.obs['pct_counts_ribo'] > 5, :]
print("Remaining cells %d"%adata.n_obs)Remaining cells 7431As you can see, a large proportion of sample covid_15 is filtered out.
5.3 Plot filtered QC
Lets plot the same QC stats once more.
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt','pct_counts_ribo', 'pct_counts_hb'], jitter=0.4, groupby = 'sample', rotation = 45)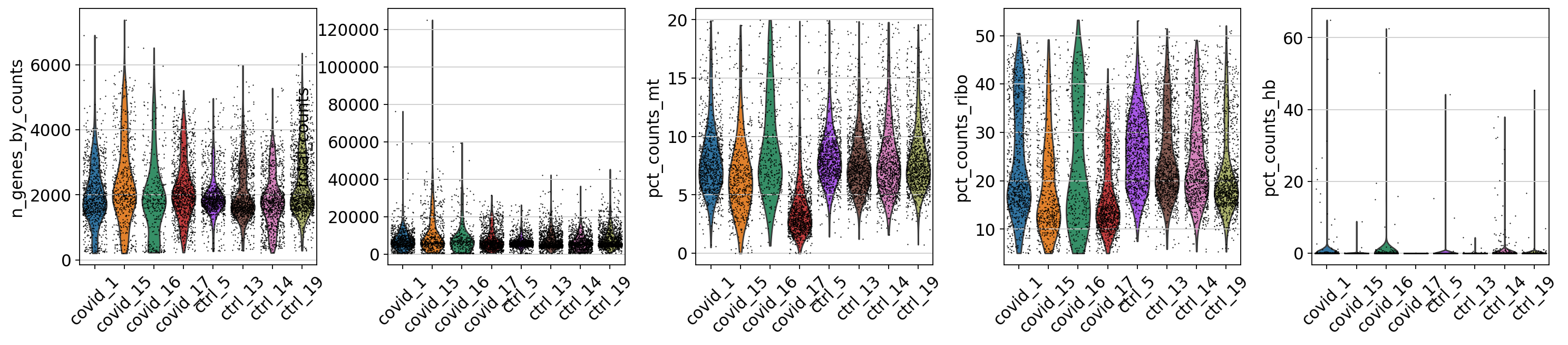
There is still quite a lot of variation in percent_mito, so it will have to be dealt with in the data analysis step. The percent_ribo values are also highly variable, but that is expected since different cell types have different proportions of ribosomal content, according to their function.
5.4 Filter genes
As the level of expression of mitochondrial and MALAT1 genes are judged as mainly technical, it can be wise to remove them from the dataset before any further analysis. In this case we will also remove the HB genes.
malat1 = adata.var_names.str.startswith('MALAT1')
# we need to redefine the mito_genes since they were first
# calculated on the full object before removing low expressed genes.
mito_genes = adata.var_names.str.startswith('MT-')
hb_genes = adata.var_names.str.contains('^HB[^(P|E|S)]')
remove = np.add(mito_genes, malat1)
remove = np.add(remove, hb_genes)
keep = np.invert(remove)
adata = adata[:,keep]
print(adata.n_obs, adata.n_vars)7431 194686 Sample sex
When working with human or animal samples, you should ideally constrain your experiments to a single sex to avoid including sex bias in the conclusions. However this may not always be possible. By looking at reads from chromosomeY (males) and XIST (X-inactive specific transcript) expression (mainly female) it is quite easy to determine per sample which sex it is. It can also be a good way to detect if there has been any mislabelling leading to the sample metadata sex not agreeing with the computational predictions.
To get choromosome information for all genes, you should ideally parse the information from the gtf file that you used in the mapping pipeline as it has the exact same annotation version/gene naming. However, it may not always be available, as in this case where we have downloaded public data. Hence, we will use biomart to fetch chromosome information.
# requires pybiomart
annot_file = 'data/covid/results/gene_annotations_pybiomart.csv'
if not os.path.exists(annot_file):
annot = sc.queries.biomart_annotations("hsapiens", ["ensembl_gene_id", "external_gene_name", "start_position", "end_position", "chromosome_name"] ).set_index("external_gene_name")
annot.to_csv(annot_file)
else:
annot = pd.read_csv(annot_file, index_col=0)Now that we have the chromosome information, we can calculate the proportion of reads that comes from chromosome Y per cell.But first we have to remove all genes in the pseudoautosmal regions of chrY that are: * chromosome:GRCh38:Y:10001 - 2781479 is shared with X: 10001 - 2781479 (PAR1) * chromosome:GRCh38:Y:56887903 - 57217415 is shared with X: 155701383 - 156030895 (PAR2)
chrY_genes = adata.var_names.intersection(annot.index[annot.chromosome_name == "Y"])
chrY_genes
par1 = [10001, 2781479]
par2 = [56887903, 57217415]
par1_genes = annot.index[(annot.chromosome_name == "Y") & (annot.start_position > par1[0]) & (annot.start_position < par1[1]) ]
par2_genes = annot.index[(annot.chromosome_name == "Y") & (annot.start_position > par2[0]) & (annot.start_position < par2[1]) ]
chrY_genes = chrY_genes.difference(par1_genes)
chrY_genes = chrY_genes.difference(par2_genes)
adata.obs['percent_chrY'] = np.sum(
adata[:, chrY_genes].X, axis=1).A1 / np.sum(adata.X, axis=1).A1 * 100Then plot XIST expression vs chrY proportion. As you can see, the samples are clearly on either side, even if some cells do not have detection of either.
# color inputs must be from either .obs or .var, so add in XIST expression to obs.
adata.obs["XIST-counts"] = adata.X[:,adata.var_names.str.match('XIST')].toarray()
sc.pl.scatter(adata, x='XIST-counts', y='percent_chrY', color="sample")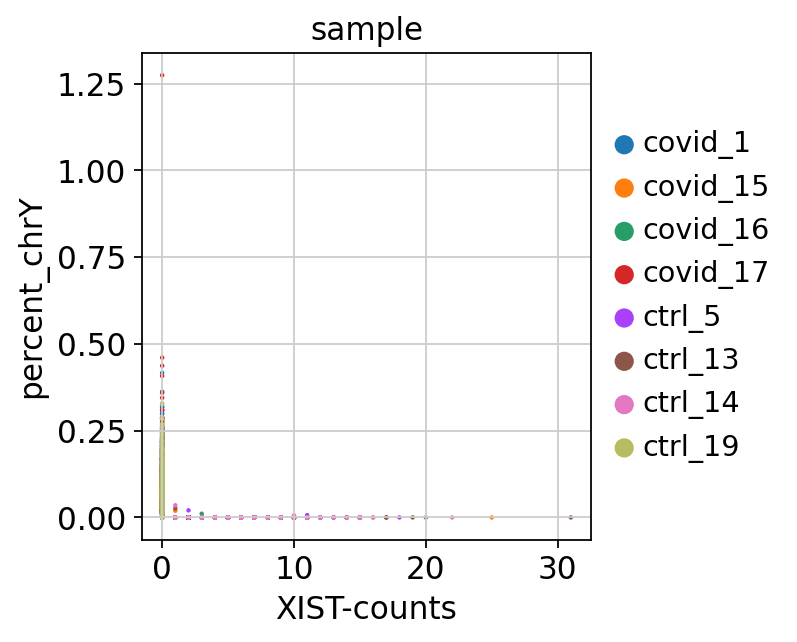
Plot as violins.
sc.pl.violin(adata, ["XIST-counts", "percent_chrY"], jitter=0.4, groupby = 'sample', rotation= 45)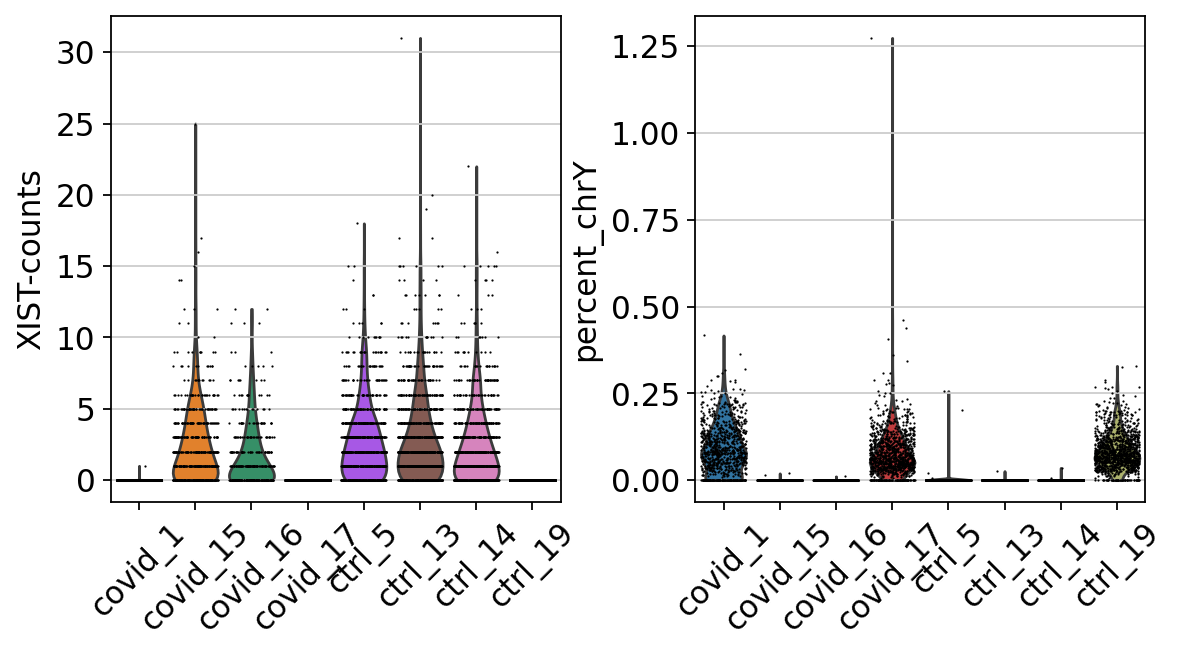
Here, we can see clearly that we have three males and five females, can you see which samples they are? Do you think this will cause any problems for downstream analysis? Discuss with your group: what would be the best way to deal with this type of sex bias?
7 Cell cycle state
We here perform cell cycle scoring. To score a gene list, the algorithm calculates the difference of mean expression of the given list and the mean expression of reference genes. To build the reference, the function randomly chooses a bunch of genes matching the distribution of the expression of the given list. Cell cycle scoring adds three slots in the metadata, a score for S phase, a score for G2M phase and the predicted cell cycle phase.
First read the file with cell cycle genes, from Regev lab and split into S and G2M phase genes. We first download the file.
path_file = os.path.join(path_results, 'regev_lab_cell_cycle_genes.txt')
if not os.path.exists(path_file):
file_url = os.path.join(path_data, "misc/regev_lab_cell_cycle_genes.txt")
subprocess.call(["curl", "-u", curl_upass, "-o", path_file, file_url ])cell_cycle_genes = [x.strip() for x in open('./data/covid/results/regev_lab_cell_cycle_genes.txt')]
print(len(cell_cycle_genes))
# Split into 2 lists
s_genes = cell_cycle_genes[:43]
g2m_genes = cell_cycle_genes[43:]
cell_cycle_genes = [x for x in cell_cycle_genes if x in adata.var_names]
print(len(cell_cycle_genes))97
94Before running cell cycle we have to normalize the data. In the scanpy object, the data slot will be overwritten with the normalized data. So first, save the raw data into the slot raw. Then run normalization, log transformation and scale the data.
# save raw counts in raw slot.
adata.raw = adata.copy()
# normalize to depth 10 000
sc.pp.normalize_total(adata, target_sum=1e4)
# logaritmize
sc.pp.log1p(adata)
# scale
sc.pp.scale(adata)normalizing counts per cell
finished (0:00:00)We here perform cell cycle scoring. The function is actually a wrapper to sc.tl.score_gene_list, which is launched twice, to score separately S and G2M phases. Both sc.tl.score_gene_list and sc.tl.score_cell_cycle_genes are a port from Seurat and are supposed to work in a very similar way. To score a gene list, the algorithm calculates the difference of mean expression of the given list and the mean expression of reference genes. To build the reference, the function randomly chooses a bunch of genes matching the distribution of the expression of the given list. Cell cycle scoring adds three slots in data, a score for S phase, a score for G2M phase and the predicted cell cycle phase.
sc.tl.score_genes_cell_cycle(adata, s_genes=s_genes, g2m_genes=g2m_genes)calculating cell cycle phase
computing score 'S_score'
WARNING: genes are not in var_names and ignored: Index(['MLF1IP'], dtype='object')
finished: added
'S_score', score of gene set (adata.obs).
774 total control genes are used. (0:00:00)
computing score 'G2M_score'
WARNING: genes are not in var_names and ignored: Index(['FAM64A', 'HN1'], dtype='object')
finished: added
'G2M_score', score of gene set (adata.obs).
772 total control genes are used. (0:00:00)
--> 'phase', cell cycle phase (adata.obs)We can now create a violin plot for the cell cycle scores as well.
sc.pl.violin(adata, ['S_score', 'G2M_score'], jitter=0.4, groupby = 'sample', rotation=45)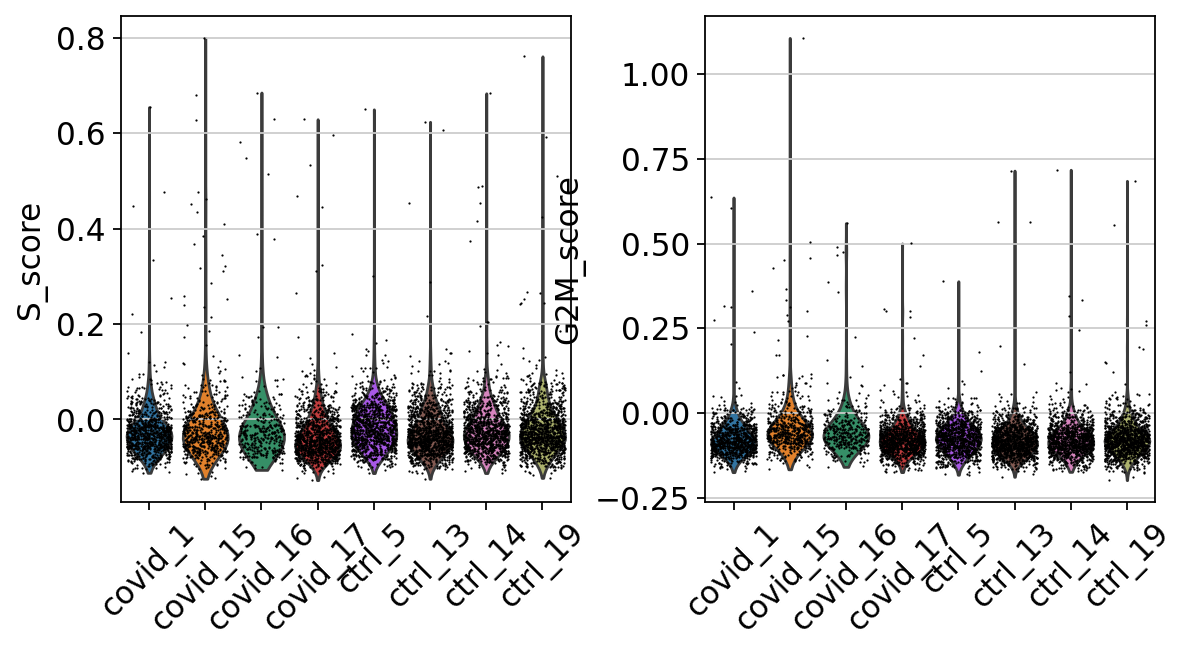
In this case it looks like we only have a few cycling cells in these datasets.
Scanpy does an automatic prediction of cell cycle phase with a default cutoff of the scores at zero. As you can see this does not fit this data very well, so be cautios with using these predictions. Instead we suggest that you look at the scores.
sc.pl.scatter(adata, x='S_score', y='G2M_score', color="phase")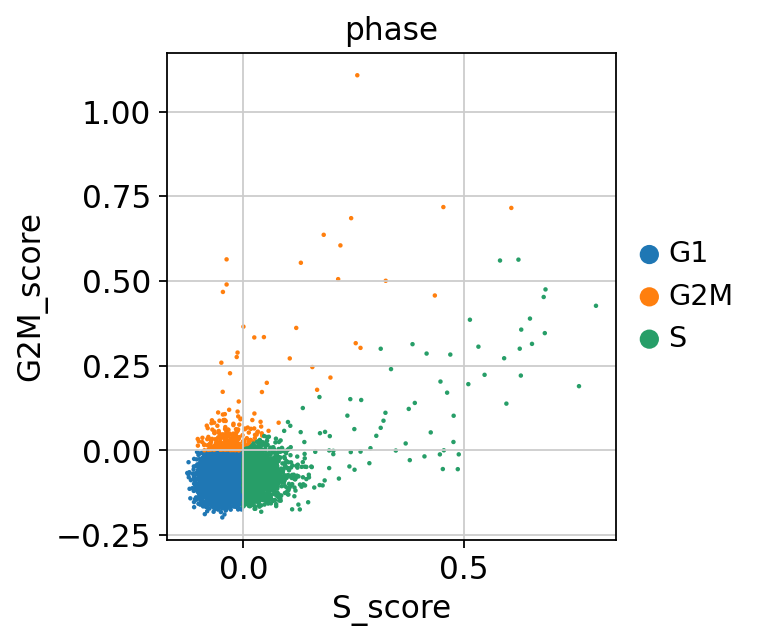
8 Predict doublets
Doublets/Multiples of cells in the same well/droplet is a common issue in scRNAseq protocols. Especially in droplet-based methods with overloading of cells. In a typical 10x experiment the proportion of doublets is linearly dependent on the amount of loaded cells. As indicated from the Chromium user guide, doublet rates are about as follows:
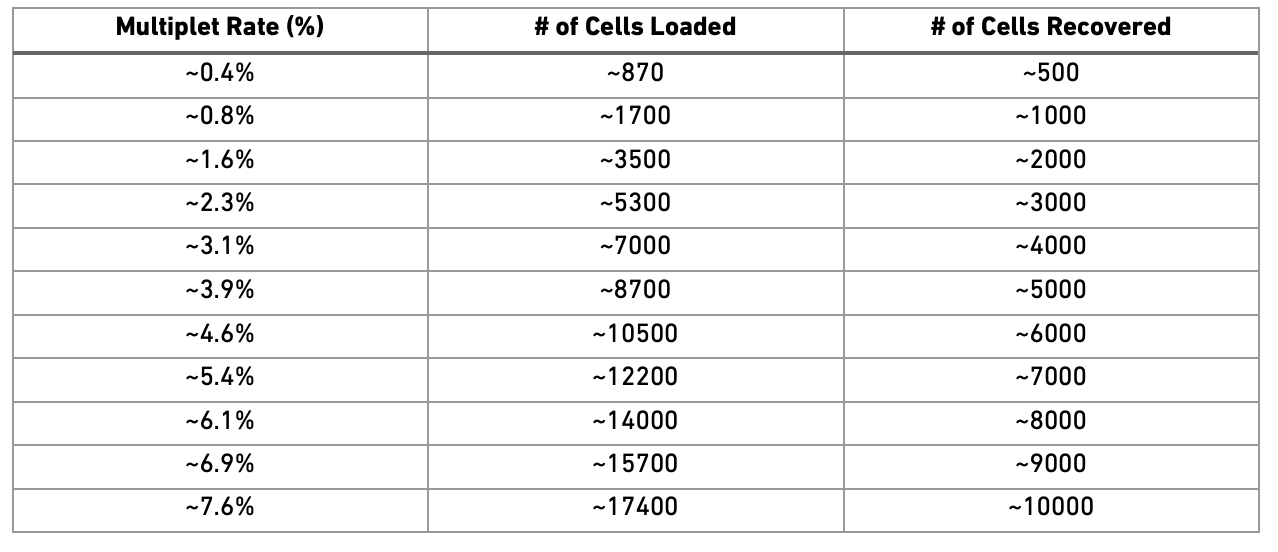
Most doublet detectors simulates doublets by merging cell counts and predicts doublets as cells that have similar embeddings as the simulated doublets. Most such packages need an assumption about the number/proportion of expected doublets in the dataset. The data you are using is subsampled, but the original datasets contained about 5 000 cells per sample, hence we can assume that they loaded about 9 000 cells and should have a doublet rate at about 4%.
For doublet detection, we will use the package Scrublet, so first we need to get the raw counts from adata.raw.X and run scrublet with that matrix. Then we add in the doublet prediction info into our anndata object.
Doublet prediction should be run for each dataset separately, so first we need to split the adata object into 8 separate objects, one per sample and then run scrublet on each of them.
import scrublet as scr
# split per batch into new objects.
batches = adata.obs['sample'].cat.categories.tolist()
alldata = {}
for batch in batches:
tmp = adata[adata.obs['sample'] == batch,]
print(batch, ":", tmp.shape[0], " cells")
scrub = scr.Scrublet(tmp.raw.X)
out = scrub.scrub_doublets(verbose=False, n_prin_comps = 20)
alldata[batch] = pd.DataFrame({'doublet_score':out[0],'predicted_doublets':out[1]},index = tmp.obs.index)
print(alldata[batch].predicted_doublets.sum(), " predicted_doublets")covid_1 : 900 cells
24 predicted_doublets
covid_15 : 599 cells
8 predicted_doublets
covid_16 : 373 cells
3 predicted_doublets
covid_17 : 1101 cells
17 predicted_doublets
ctrl_5 : 1052 cells
35 predicted_doublets
ctrl_13 : 1173 cells
52 predicted_doublets
ctrl_14 : 1063 cells
33 predicted_doublets
ctrl_19 : 1170 cells
37 predicted_doublets# add predictions to the adata object.
scrub_pred = pd.concat(alldata.values())
adata.obs['doublet_scores'] = scrub_pred['doublet_score']
adata.obs['predicted_doublets'] = scrub_pred['predicted_doublets']
sum(adata.obs['predicted_doublets'])209We should expect that two cells have more detected genes than a single cell, lets check if our predicted doublets also have more detected genes in general.
# add in column with singlet/doublet instead of True/Fals
%matplotlib inline
adata.obs['doublet_info'] = adata.obs["predicted_doublets"].astype(str)
sc.pl.violin(adata, 'n_genes_by_counts', jitter=0.4, groupby = 'doublet_info', rotation=45)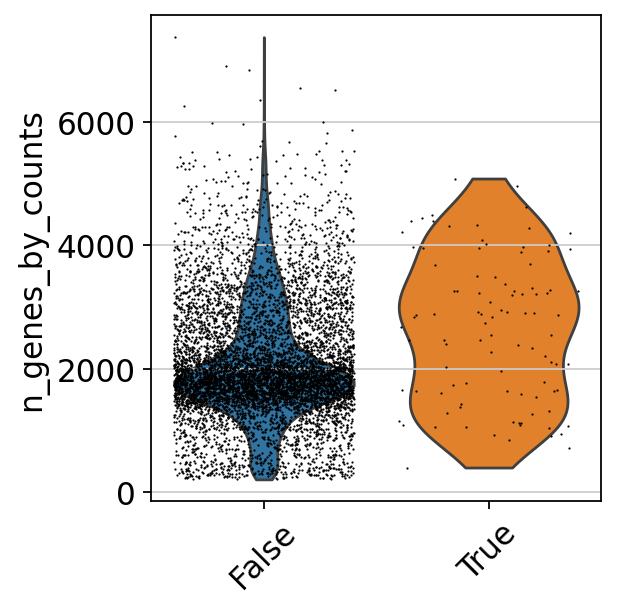
Now, lets run PCA and UMAP and plot doublet scores onto UMAP to check the doublet predictions. We will go through these steps in more detail in the later exercises.
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
adata = adata[:, adata.var.highly_variable]
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
sc.pp.scale(adata, max_value=10)
sc.pp.pca(adata, svd_solver='arpack', random_state=573)
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40, random_state=159)
sc.tl.umap(adata, random_state=331)
sc.pl.umap(adata, color=['doublet_scores','doublet_info','sample'])extracting highly variable genes
finished (0:00:01)
--> added
'highly_variable', boolean vector (adata.var)
'means', float vector (adata.var)
'dispersions', float vector (adata.var)
'dispersions_norm', float vector (adata.var)
regressing out ['total_counts', 'pct_counts_mt']
finished (0:00:01)
computing PCA
with n_comps=50
finished (0:00:01)
computing neighbors
using 'X_pca' with n_pcs = 40
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:06)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm)
'umap', UMAP parameters (adata.uns) (0:00:09)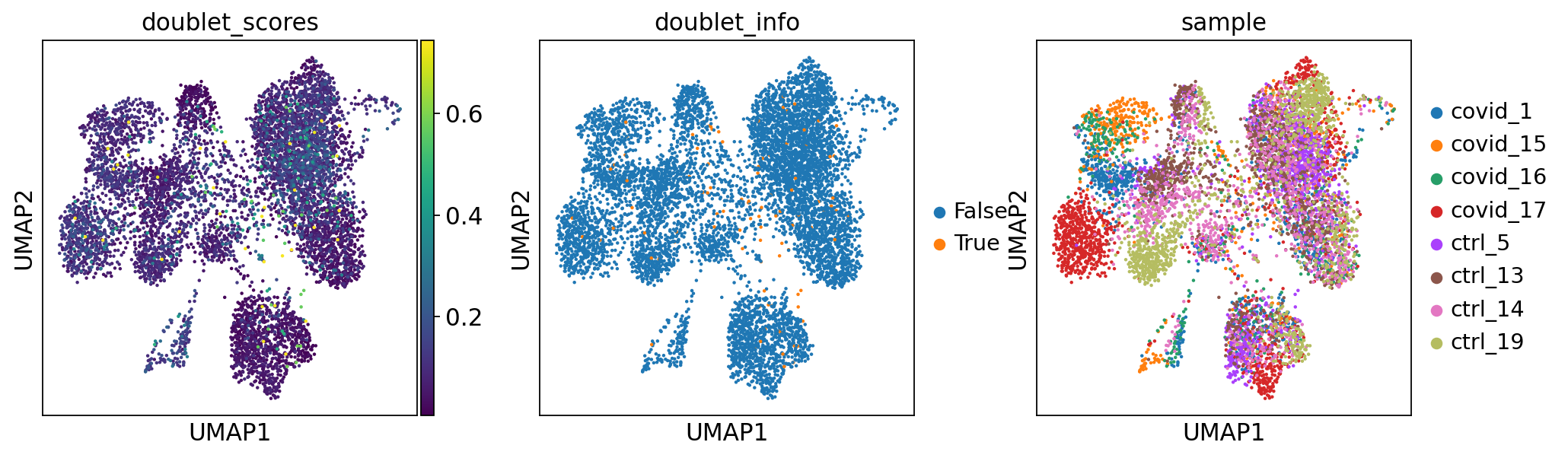
Now, lets remove all predicted doublets from our data.
# also revert back to the raw counts as the main matrix in adata
adata = adata.raw.to_adata()
adata = adata[adata.obs['doublet_info'] == 'False',:]
print(adata.shape)(7222, 19468)To summarize, lets check how many cells we have removed per sample, we started with 1500 cells per sample. Looking back at the intitial QC plots does it make sense that some samples have much fewer cells now?
#| label: view-data
adata.obs["sample"].value_counts()
NoteDiscuss
Why is it important to predict doublets in the different samples separately? In which situations would this be more/less important?
9 Save data
Finally, lets save the QC-filtered data for further analysis. Create output directory data/covid/results and save data to that folder. This will be used in downstream labs.
adata.write_h5ad('data/covid/results/scanpy_covid_qc.h5ad')10 Session info
Click here
sc.logging.print_versions()Dependencies
| Dependency | Version |
|---|---|
| statsmodels | 0.14.6 |
| parso | 0.8.6 |
| debugpy | 1.8.20 |
| ipykernel | 7.2.0 |
| asttokens | 3.0.1 |
| decorator | 5.2.1 |
| numba | 0.63.1 |
| python-dateutil | 2.9.0.post0 |
| xarray | 2026.2.0 |
| jedi | 0.19.2 |
| matplotlib | 3.10.8 |
| psutil | 7.2.2 |
| fast-array-utils | 1.3.1 |
| seaborn | 0.13.2 |
| jupyter_core | 5.9.1 |
| donfig | 0.8.1.post1 |
| pytz | 2025.2 |
| tqdm | 4.67.3 |
| setuptools | 82.0.0 |
| traitlets | 5.14.3 |
| cycler | 0.12.1 |
| h5py | 3.15.1 |
| kiwisolver | 1.4.9 |
| ipython | 9.10.0 |
| backports.zstd | 1.3.0 |
| pynndescent | 0.5.13 |
| pyzmq | 27.1.0 |
| lazy_loader | 0.4 |
| typing_extensions | 4.15.0 |
| PyYAML | 6.0.3 |
| numcodecs | 0.16.5 |
| msgpack | 1.1.2 |
| scikit-learn | 1.6.1 |
| wcwidth | 0.6.0 |
| google-crc32c | 1.8.0 |
| matplotlib-inline | 0.2.1 |
| platformdirs | 4.9.2 |
| scikit-image | 0.26.0 |
| pillow | 12.1.1 |
| session-info2 | 0.4 |
| sparse | 0.18.0 |
| jupyter_client | 8.8.0 |
| patsy | 1.0.2 |
| Pygments | 2.19.2 |
| legacy-api-wrap | 1.5 |
| six | 1.17.0 |
| joblib | 1.5.3 |
| comm | 0.2.3 |
| pyparsing | 3.3.2 |
| llvmlite | 0.46.0 |
| prompt_toolkit | 3.0.52 |
| umap-learn | 0.5.11 |
| natsort | 8.4.0 |
| packaging | 26.0 |
| charset-normalizer | 3.4.4 |
| executing | 2.2.1 |
| threadpoolctl | 3.6.0 |
| scipy | 1.16.3 |
| pure_eval | 0.2.3 |
| zarr | 3.1.5 |
| fsspec | 2026.2.0 |
| stack_data | 0.6.3 |
| annoy | 1.17.3 |
| tornado | 6.5.3 |
Copyable Markdown
| Package | Version | | -------- | ------- | | numpy | 2.3.5 | | pandas | 2.3.3 | | scanpy | 1.12 | | anndata | 0.12.10 | | scrublet | 0.2.3 | | Dependency | Version | | ------------------ | ----------- | | statsmodels | 0.14.6 | | parso | 0.8.6 | | debugpy | 1.8.20 | | ipykernel | 7.2.0 | | asttokens | 3.0.1 | | decorator | 5.2.1 | | numba | 0.63.1 | | python-dateutil | 2.9.0.post0 | | xarray | 2026.2.0 | | jedi | 0.19.2 | | matplotlib | 3.10.8 | | psutil | 7.2.2 | | fast-array-utils | 1.3.1 | | seaborn | 0.13.2 | | jupyter_core | 5.9.1 | | donfig | 0.8.1.post1 | | pytz | 2025.2 | | tqdm | 4.67.3 | | setuptools | 82.0.0 | | traitlets | 5.14.3 | | cycler | 0.12.1 | | h5py | 3.15.1 | | kiwisolver | 1.4.9 | | ipython | 9.10.0 | | backports.zstd | 1.3.0 | | pynndescent | 0.5.13 | | pyzmq | 27.1.0 | | lazy_loader | 0.4 | | typing_extensions | 4.15.0 | | PyYAML | 6.0.3 | | numcodecs | 0.16.5 | | msgpack | 1.1.2 | | scikit-learn | 1.6.1 | | wcwidth | 0.6.0 | | google-crc32c | 1.8.0 | | matplotlib-inline | 0.2.1 | | platformdirs | 4.9.2 | | scikit-image | 0.26.0 | | pillow | 12.1.1 | | session-info2 | 0.4 | | sparse | 0.18.0 | | jupyter_client | 8.8.0 | | patsy | 1.0.2 | | Pygments | 2.19.2 | | legacy-api-wrap | 1.5 | | six | 1.17.0 | | joblib | 1.5.3 | | comm | 0.2.3 | | pyparsing | 3.3.2 | | llvmlite | 0.46.0 | | prompt_toolkit | 3.0.52 | | umap-learn | 0.5.11 | | natsort | 8.4.0 | | packaging | 26.0 | | charset-normalizer | 3.4.4 | | executing | 2.2.1 | | threadpoolctl | 3.6.0 | | scipy | 1.16.3 | | pure_eval | 0.2.3 | | zarr | 3.1.5 | | fsspec | 2026.2.0 | | stack_data | 0.6.3 | | annoy | 1.17.3 | | tornado | 6.5.3 | | Component | Info | | --------- | ------------------------------------------------------------------------------ | | Python | 3.12.12 | packaged by conda-forge | (main, Jan 26 2026, 23:51:32) [GCC 14.3.0] | | OS | Linux-6.12.5-linuxkit-x86_64-with-glibc2.35 | | CPU | 16 logical CPU cores, x86_64 | | GPU | No GPU found | | Updated | 2026-04-17 10:19 |