Making reproducible workflows with 
Why do we need workflow managers?
- As projects grow or age, it becomes increasingly difficult to keep track of all the parts and how they fit together.
Why do we need workflow managers?
- As projects grow or age, it becomes increasingly difficult to keep track of all the parts and how they fit together.
Why do we need workflow managers?
- As projects grow or age, it becomes increasingly difficult to keep track of all the parts and how they fit together.
Why do we need workflow managers?
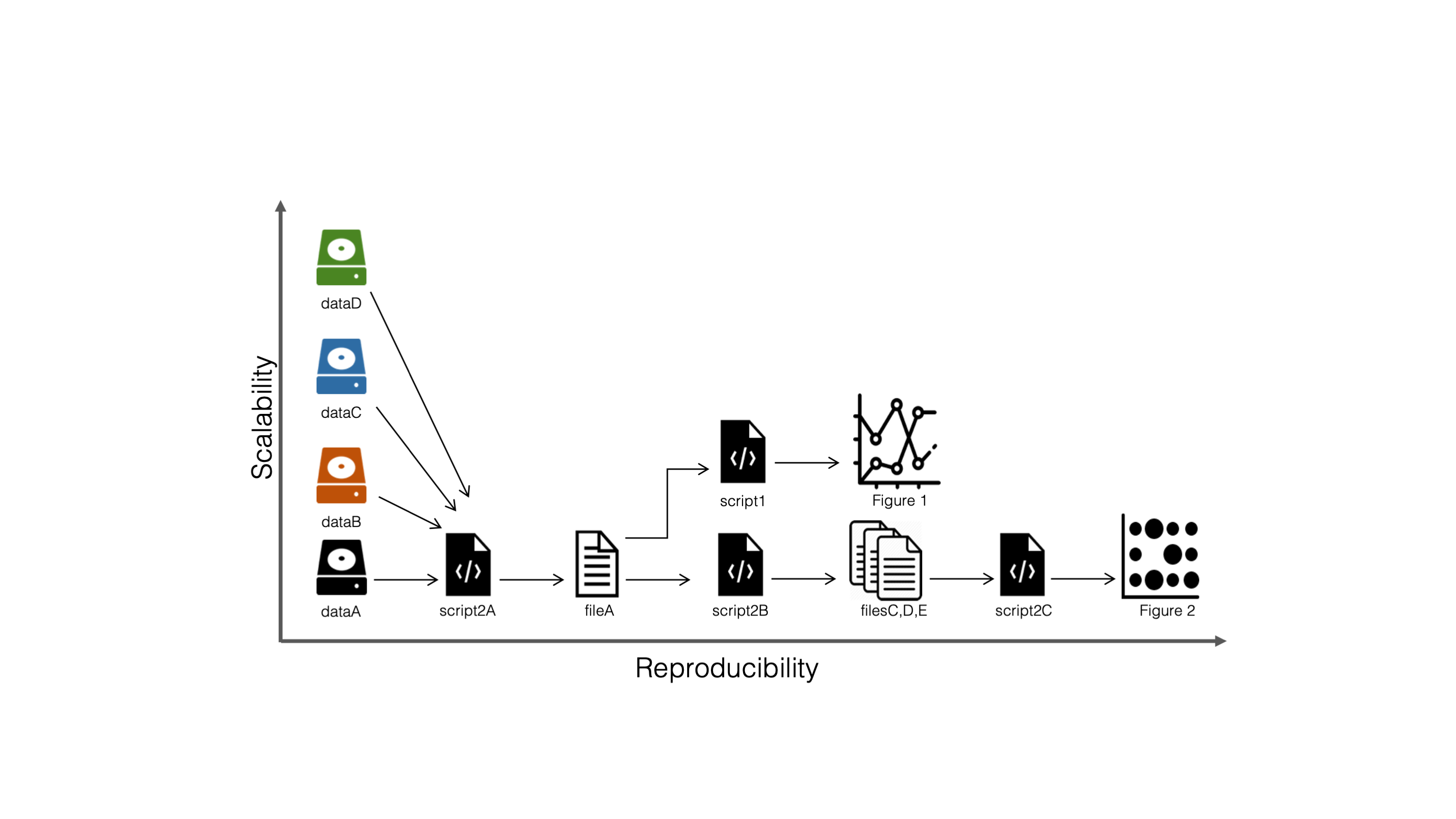
- A workflow manager helps you scale up both in complexity and dataset size
Workflow managers for bioinformatics
Most common
- Snakemake
- Nextflow
Others
- Makeflow
- Bpipe
- Pachyderm
Snakemake workflows
- Automatically track input/output file dependencies
- Are built from rules
- Are generalized with wildcards
- Use a Python-based definition language
- Easily scale from laptops to HPC clusters
Reproducible…
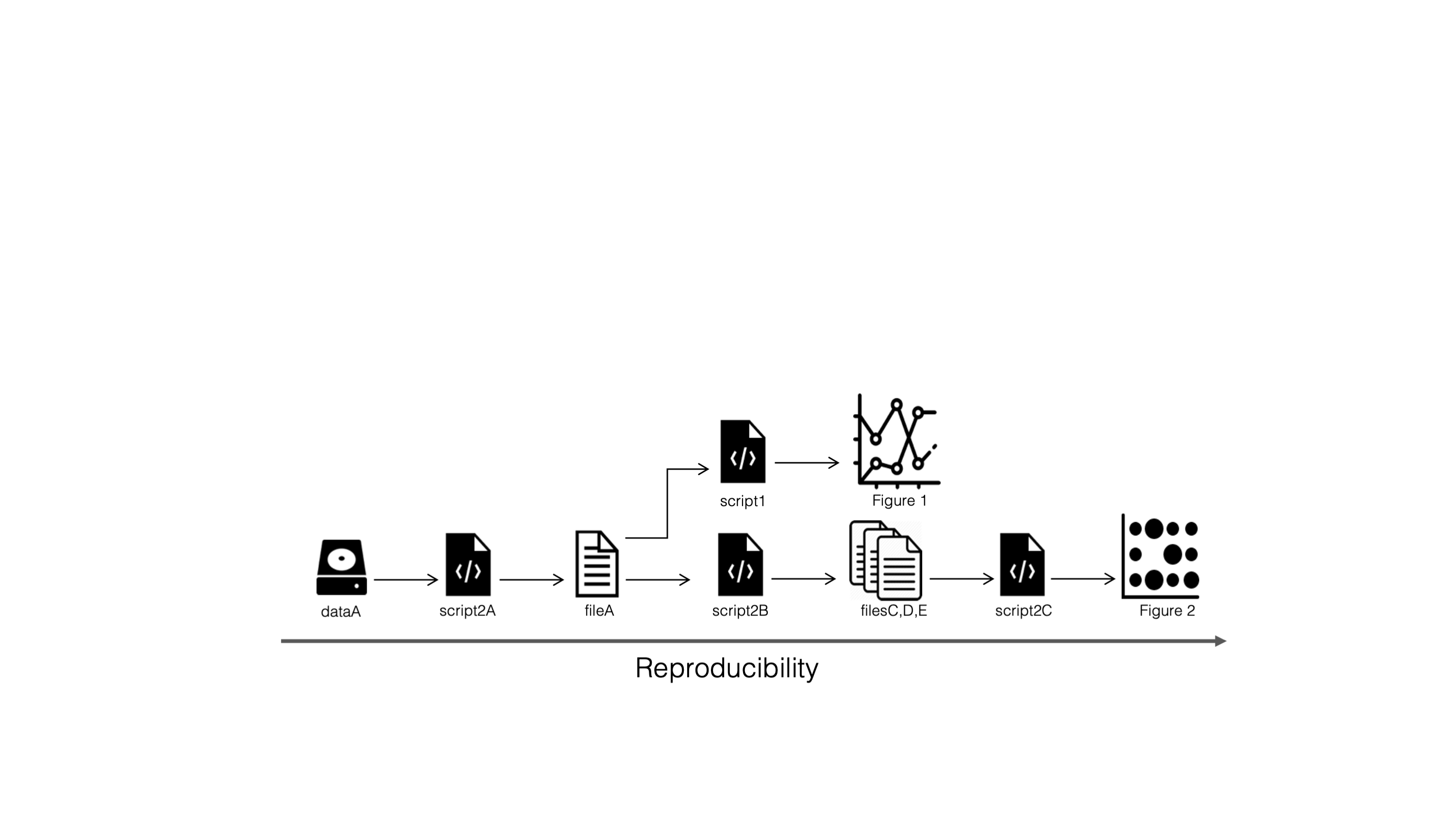
…and scalable workflows
Example: sequence trimming
Goal: Create workflow to trim and compress FASTQ files
Example: sequence trimming
Using a bash-script:
trimfastq.sh
for input in *.fastq
do
sample=$(echo ${input} | sed 's/.fastq//')
# 1. Trim fastq file (trim 5 bp from left, 10 bp from right)
seqtk trimfq -b 5 -e 10 $input > ${sample}.trimmed.fastq
# 2. Compress fastq file
gzip -c ${sample}.trimmed.fastq > ${sample}.trimmed.fastq.gz
# 3. Remove intermediate files
rm ${sample}.trimmed.fastq
doneExample: sequence trimming
Using Snakemake rules:
Snakefile
$ snakemake -c 1 a.trimmed.fastq.gz b.trimmed.fastq.gz
Building DAG of jobs...
Using shell: /bin/bash
Provided cores: 1 (use --cores to define parallelism)
Rules claiming more threads will be scaled down.
Job stats:
job count
---------- -------
gzip 2
trim_fastq 2
total 4
Select jobs to execute...
Execute 1 jobs...
[Tue Nov 19 23:09:00 2024]
localrule trim_fastq:
input: b.fastq
output: b.trimmed.fastq
jobid: 3
reason: Missing output files: b.trimmed.fastq
wildcards: sample=b
resources: tmpdir=/var/folders/wb/jf9h8kw11b734gd98s6174rm0000gp/T
[Tue Nov 19 23:09:01 2024]
Finished job 3.
1 of 4 steps (25%) done
Select jobs to execute...
Execute 1 jobs...
[Tue Nov 19 23:09:01 2024]
localrule gzip:
input: b.trimmed.fastq
output: b.trimmed.fastq.gz
jobid: 2
reason: Missing output files: b.trimmed.fastq.gz; Input files updated by another job: b.trimmed.fastq
wildcards: sample=b
resources: tmpdir=/var/folders/wb/jf9h8kw11b734gd98s6174rm0000gp/T
[Tue Nov 19 23:09:02 2024]
Finished job 2.
2 of 4 steps (50%) done
Removing temporary output b.trimmed.fastq.
Select jobs to execute...
Execute 1 jobs...
[Tue Nov 19 23:09:02 2024]
localrule trim_fastq:
input: a.fastq
output: a.trimmed.fastq
jobid: 1
reason: Missing output files: a.trimmed.fastq
wildcards: sample=a
resources: tmpdir=/var/folders/wb/jf9h8kw11b734gd98s6174rm0000gp/T
[Tue Nov 19 23:09:02 2024]
Finished job 1.
3 of 4 steps (75%) done
Select jobs to execute...
Execute 1 jobs...
[Tue Nov 19 23:09:02 2024]
localrule gzip:
input: a.trimmed.fastq
output: a.trimmed.fastq.gz
jobid: 0
reason: Missing output files: a.trimmed.fastq.gz; Input files updated by another job: a.trimmed.fastq
wildcards: sample=a
resources: tmpdir=/var/folders/wb/jf9h8kw11b734gd98s6174rm0000gp/T
[Tue Nov 19 23:09:03 2024]
Finished job 0.
4 of 4 steps (100%) done
Removing temporary output a.trimmed.fastq.
Complete log: .snakemake/log/2024-11-19T230900.634412.snakemake.logGetting into the Snakemake mindset
From the Snakemake documentation:
“A Snakemake workflow is defined by specifying rules in a Snakefile.”
“Rules decompose the workflow into small steps.”
“Snakemake automatically determines the dependencies between the rules by matching file names.”
- By themselves, rules only define what files can be generated
- By themselves, rules only define what files can be generated
- The actual rules to run are determined automatically from the files you want, so called targets
$ snakemake -c 1 a.trimmed.fastq.gz b.trimmed.fastq.gz- By themselves, rules only define what files can be generated
- The actual rules to run are determined automatically from the files you want, so-called targets
$ snakemake -c 1 a.trimmed.fastq.gz- By themselves, rules only define what files can be generated
- The actual rules to run are determined automatically from the files you want, so called targets
- It can therefore be helpful to think of Snakemake workflows in a bottom-up manner, starting with the output
- By themselves, rules only define what files can be generated
- The actual rules to run are determined automatically from the files you want, so called targets
- It can therefore be helpful to think of Snakemake workflows in a bottom-up manner, starting with the output
- If no target is passed at the command line, Snakemake will use the first defined rule in the Snakefile as a target
How does Snakemake keep track of what files to generate?
Example from the practical tutorial
- The tutorial contains a workflow to download and map RNA-seq reads against a reference genome.
- Here we ask for results/supplementary.html, which is a Quarto report generated by the rule
make_supplementary:
$ snakemake -c 1 results/supplementary.html- If the timestamp of a file upstream in the workflow is updated…
$ touch results/bowtie2/NCTC8325.1.bt2- Snakemake detects a file change and only reruns the necessary rules.
$ snakemake -c 1 results/supplementary.htmlAnatomy of a Snakemake rule
- Rules are typically named and have input and/or output directives
- Logfiles help with debugging and leave a “paper trail”
- Params can be used to pass on settings
- The
threadsdirective specify maximum number of threads for a rule - You can also define
resourcessuch as disk/memory requirements and runtime
rule trim_fastq:
output: temp("{sample}.trimmed.fastq")
input: "{sample}.fastq"
log: "logs/{sample}.trim_fastq.log"
params:
leftTrim=5,
rightTrim=10
threads: 8
resources:
mem_mb=64,
runtime=120
shell:
"""
seqtk trimfq -t {threads} -b {params.leftTrim} -e {params.rightTrim} {input} > {output} 2> {log}
"""- Rules can be executed in separate software environments using either the
condaorcontainerdirective
rule trim_fastq:
output: temp("{sample}.trimmed.fastq")
input: "{sample}.fastq"
log: "logs/{sample}.trim_fastq.log"
params:
leftTrim=5,
rightTrim=10
threads: 8
resources:
mem_mb=64,
runtime=120
conda: "envs/seqtk.yaml"
container: "docker://quay.io/biocontainers/seqtk"
shell:
"""
seqtk trimfq -t {threads} -b {params.leftTrim} -e {params.rightTrim} {input} > {output} 2> {log}
"""- Rules can be executed in separate software environments using either the
condaorcontainerdirective
See more in the Snakemake documentation
https://snakemake.readthedocs.io/en/stable/snakefiles/rules.html
Snakemake commandline
- Generate the output of the first rule in Snakefile
Snakemake commandline
- Run the workflow in dry mode and print shell commands
Snakemake commandline
- Execute the workflow with 8 cores
Snakemake commandline
- Specify a configuration file
Snakemake commandline
- Run rules with specific conda environments or Docker/Apptainer containers
Questions?

