Making reproducible workflows with 
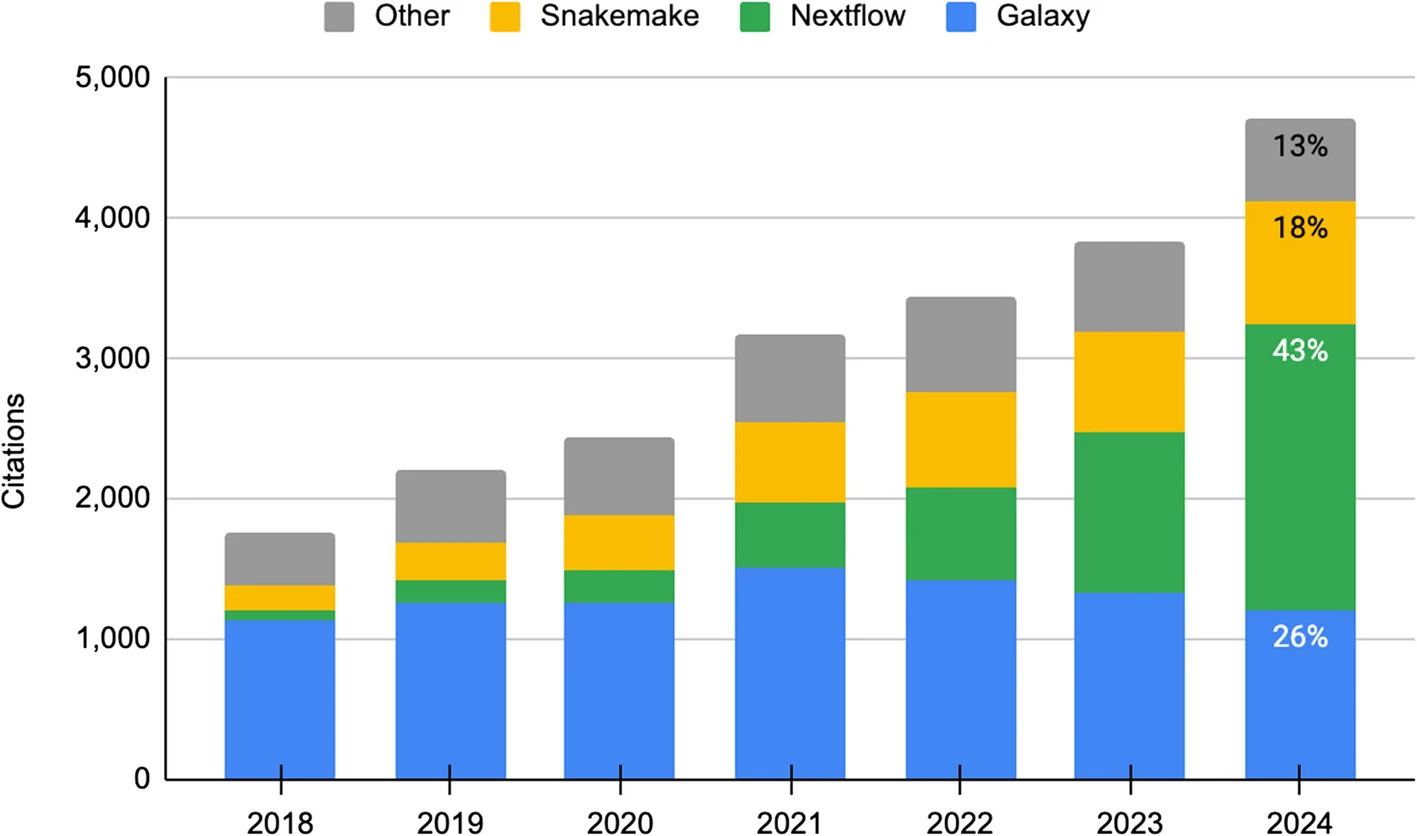
Nextflow adoption
Nextflow has grown rapidly since its creation, and is the most commonly used workflow manager in bioinformatics since 2024. 1
Concepts and nomenclature
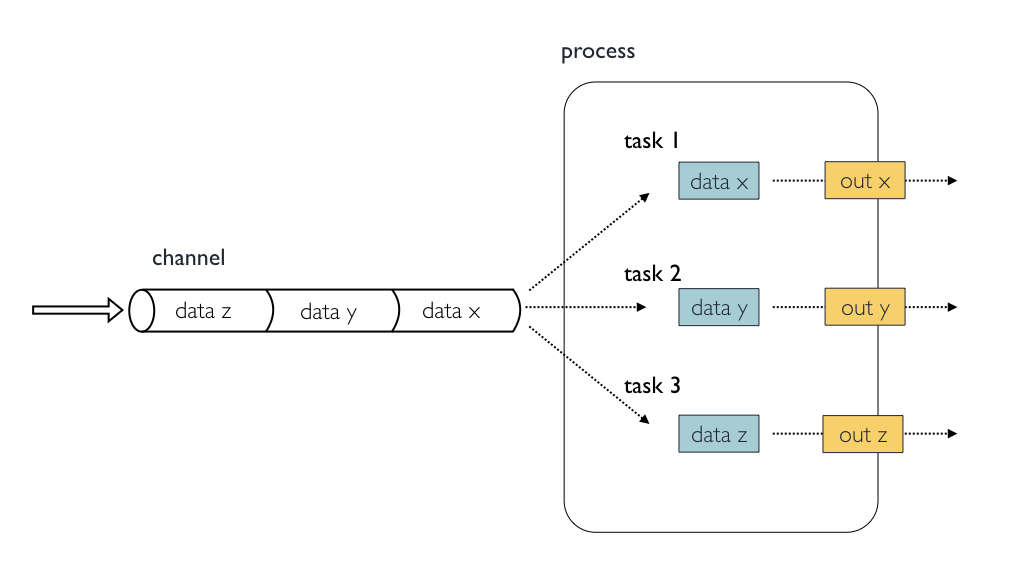
- Channels contain data, e.g. input files
- Processes run some kind of code, e.g. a script or a program
- Tasks are instances of a process, one per process input
- Each task is run in its own, isolated sub-directory
Questions?

