Bulk RNASeq Analysis
29-Nov-2024

What is RNA?
![]()
- The transcriptome is spatially and temporally dynamic
- Data comes from functional units (coding regions)
- Only a tiny fraction of the genome
Workflow


Conesa et al. (2016)
Experimental design
- Biological replicates: 6 - 12 Schurch et al. (2016)
- Sample size estimation Hart et al. (2013)
- Power analysis rnaseq-power web app, Zhao et al. (2018)
- Balanced design to avoid batch effects
- RIN values have strong effect Gallego Romero et al. (2014)

Library & Sequencing


- polyA selection / Ribosomal RNA depletion
- Single-end / Paired-end
Workflow • DGE

Read QC
- Number of reads
- Per base sequence quality
- Per sequence quality score
- Per base sequence content
- Per sequence GC content
- Per base N content
- Sequence length distribution
- Sequence duplication levels
- Overrepresented sequences
- Adapter content
- Kmer content
FastQC, MultiQC, https://sequencing.qcfail.com/


FastQC
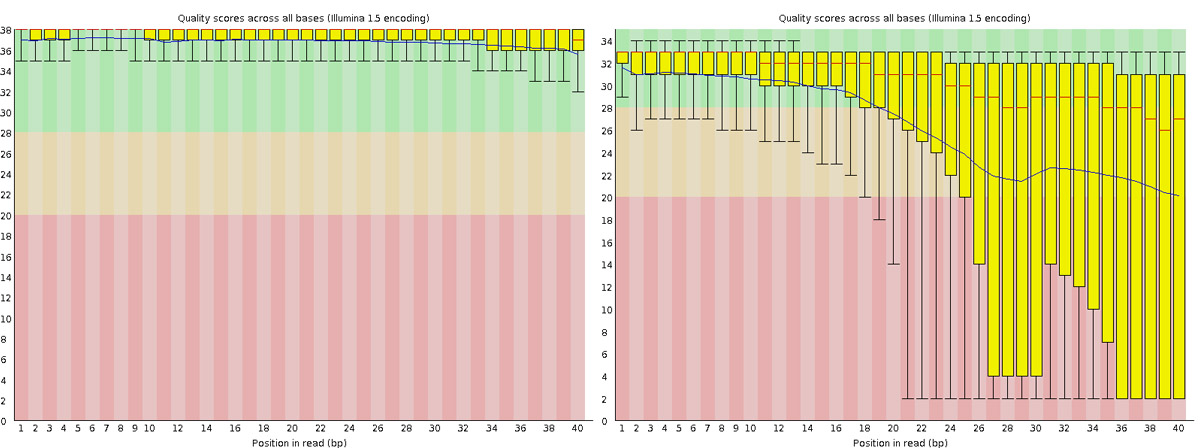
Good quality

Poor quality
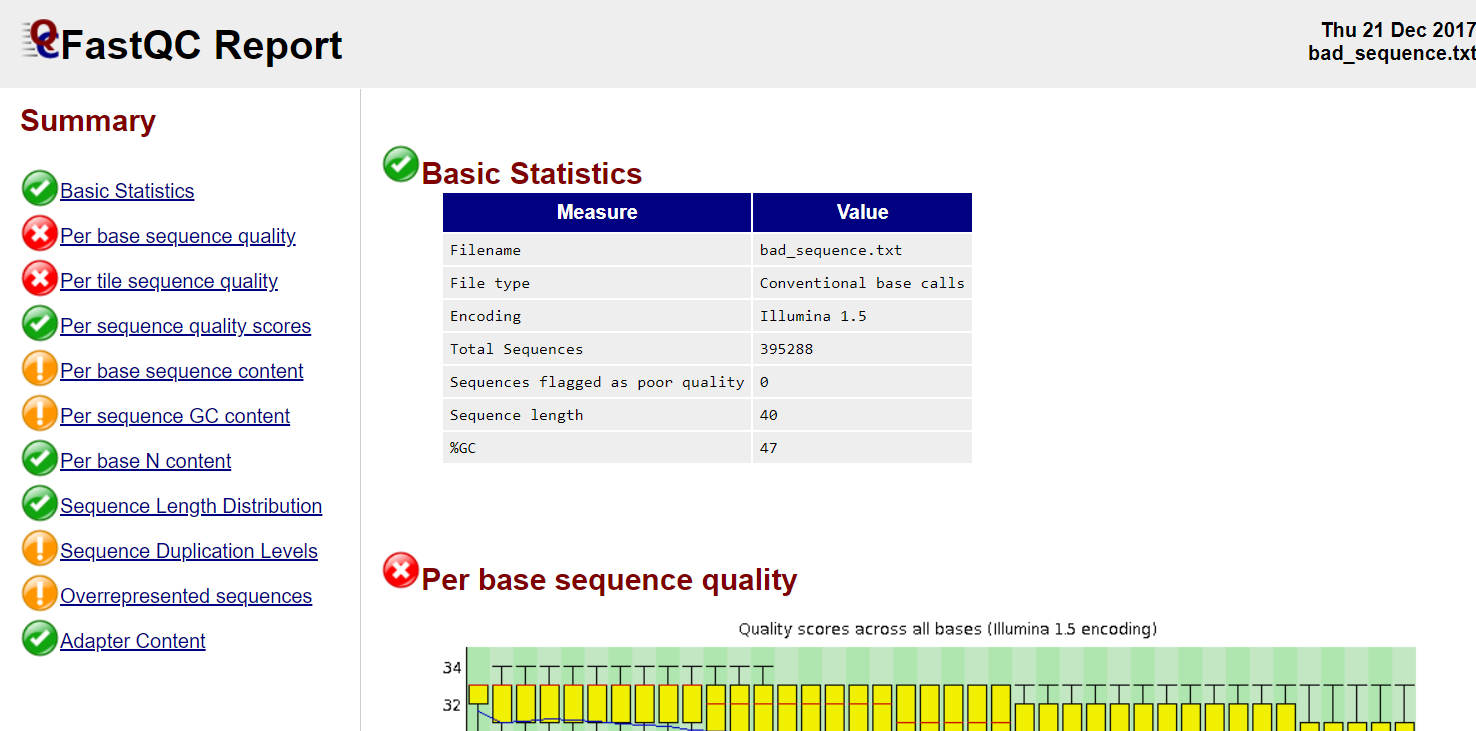
Read QC • PBSQ, PSQS
Per base sequence quality
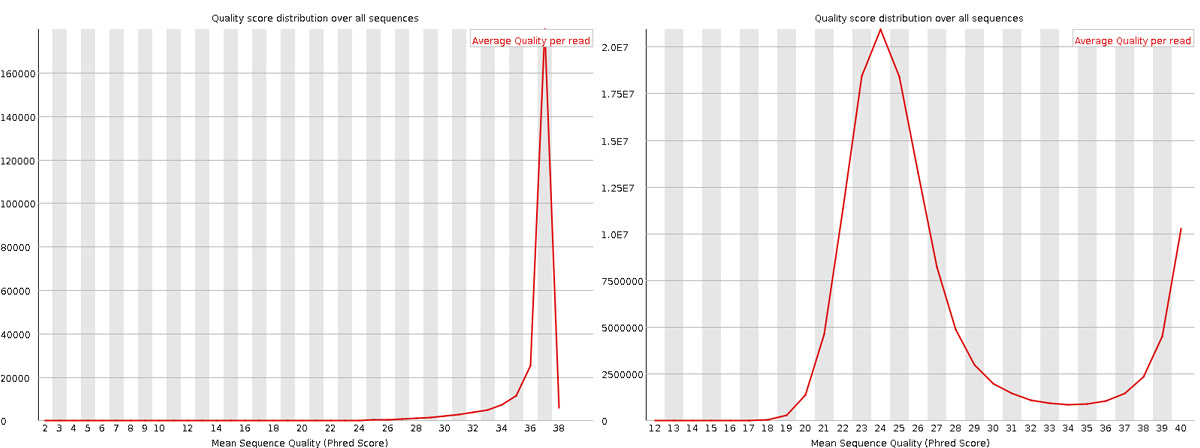
Per sequence quality scores
Trimming
- Trimming reads to remove adapter/readthrough or low quality bases
- Related options are hard clipping, filtering reads
- Sliding window trimming
- Filter by min/max read length
- Remove reads less than ~18nt
- Demultiplexing/Splitting
When to avoid trimming?
- Read trimming may not always be necessary Liao & Shi (2020)
- Fixed read length may sometimes be more important
- Expected insert size distribution may be more important for assemblers

Mapping

- Aligning reads back to a reference sequence
- Mapping to genome vs transcriptome
- Splice-aware alignment (genome) (STAR, HISAT2 etc)
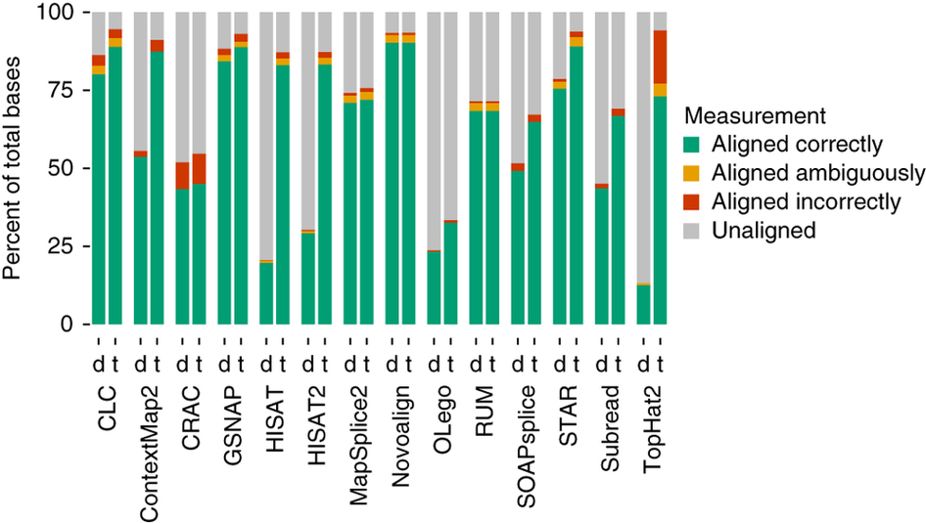
Aligners • Metrics
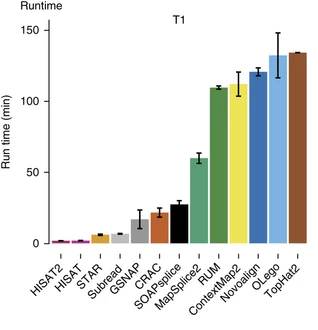
Baruzzo et al. (2017)

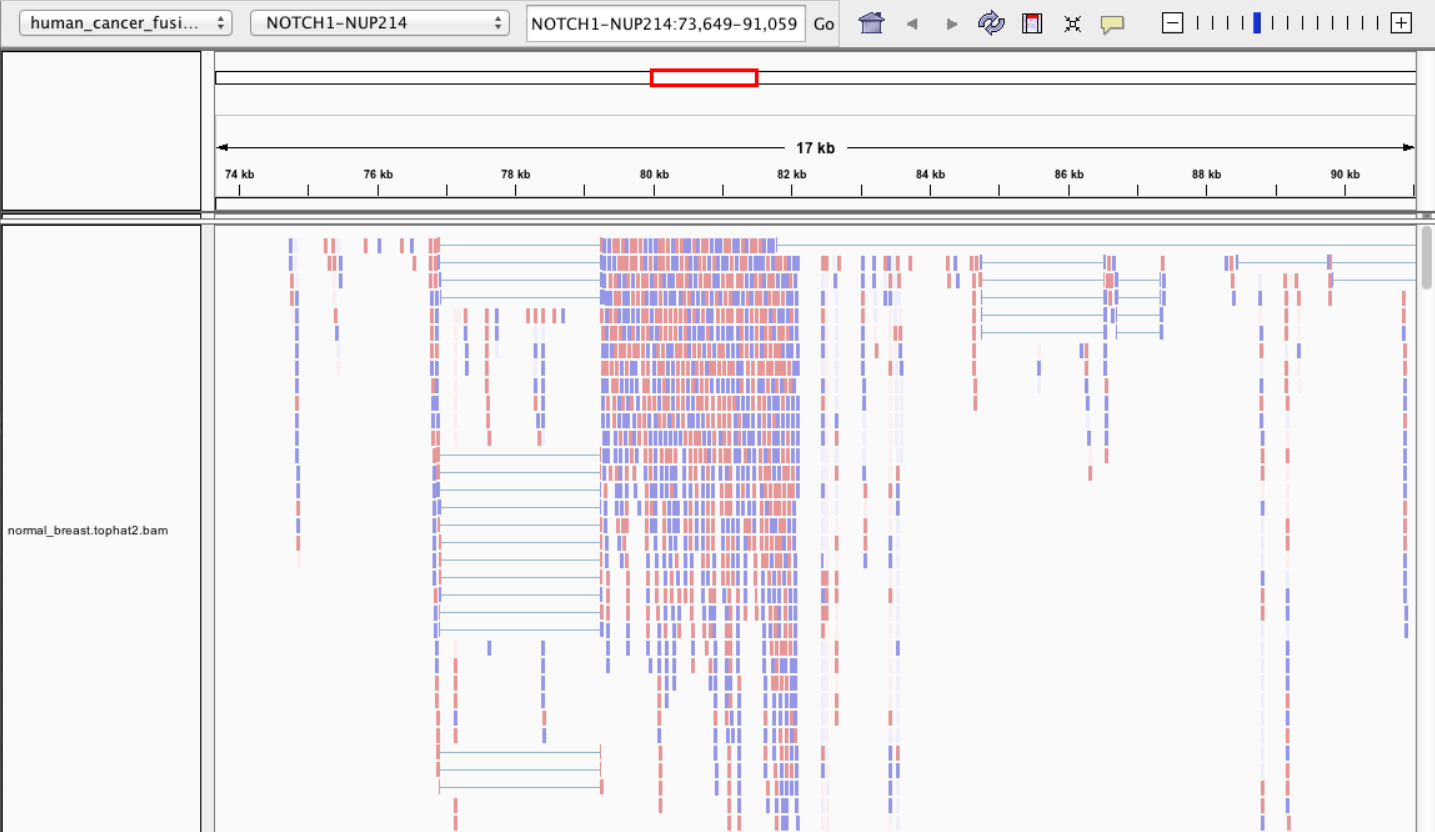
Visualisation • IGV
Visualisation • tview
samtools tview alignment.bam genome.fasta

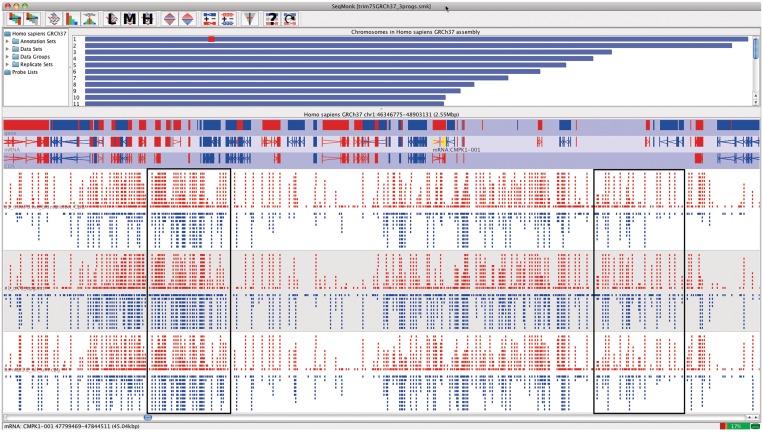
Visualisation • SeqMonk
Alignment QC • STAR Log
MultiQC can be used to summarise and plot STAR log files.

Alignment QC • Features
QoRTs was run on all samples and summarised using MultiQC.

Alignment QC • QoRTs
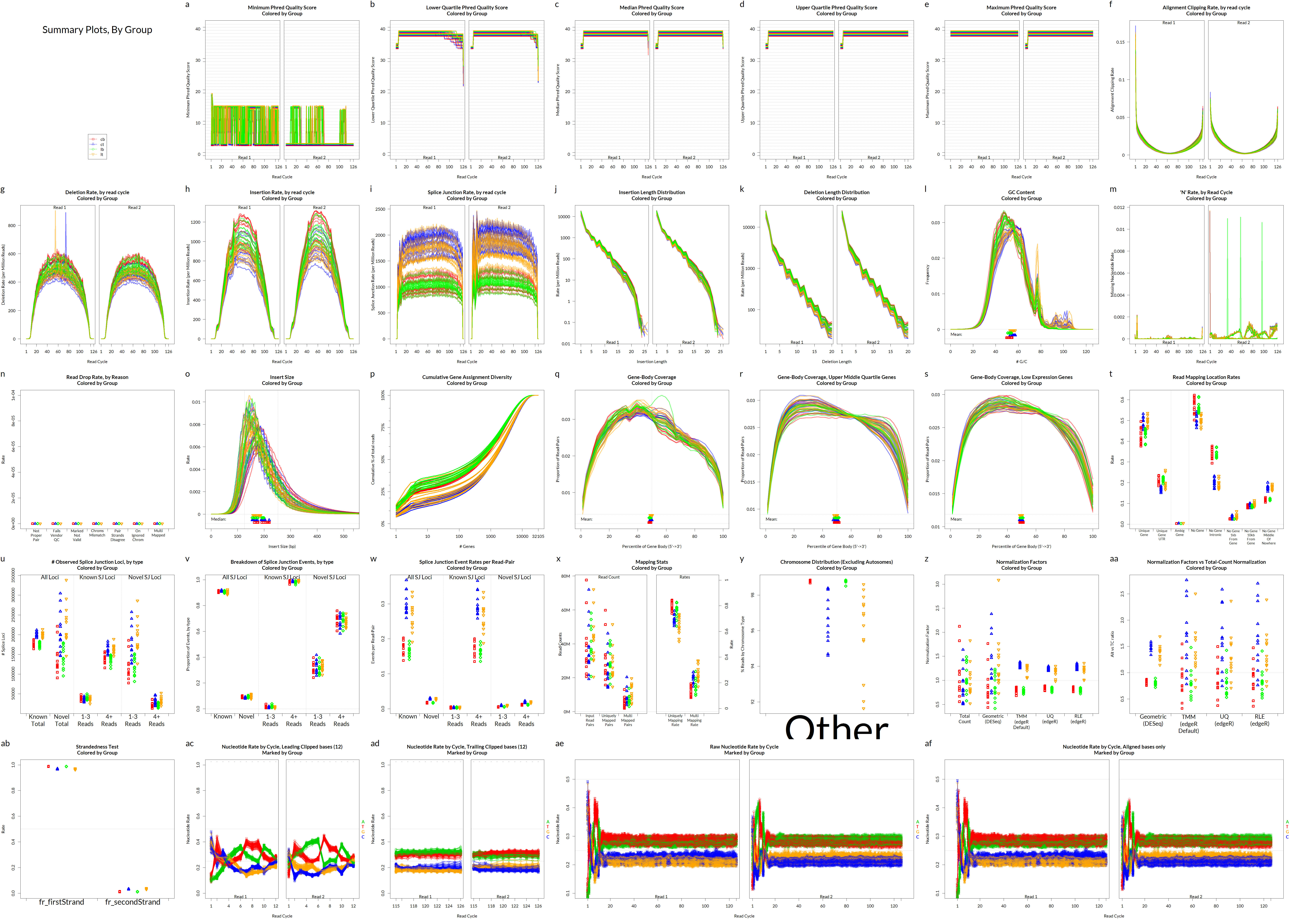
Alignment QC • Examples
Read mapping profile 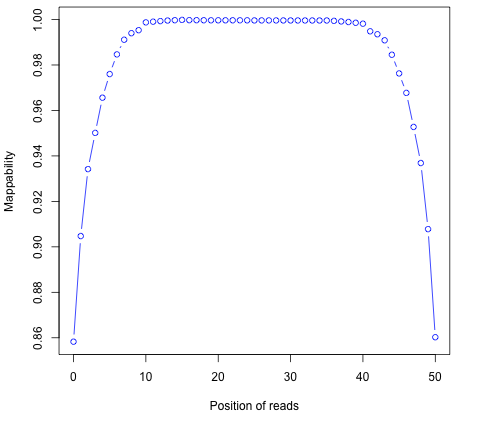
Gene body coverage 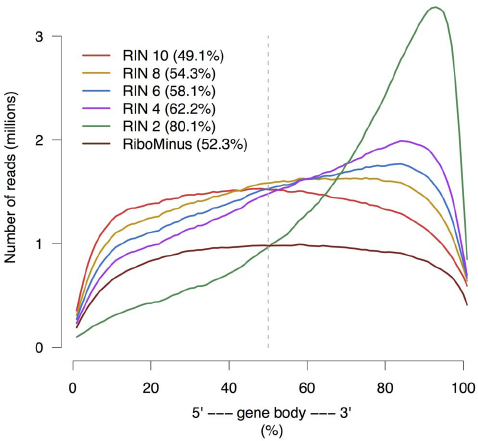
Sigurgeirsson et al. (2014)
Alignment QC • Examples
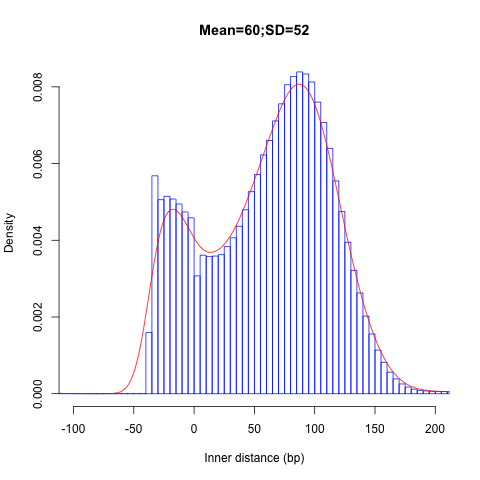
Insert size
Saturation curve
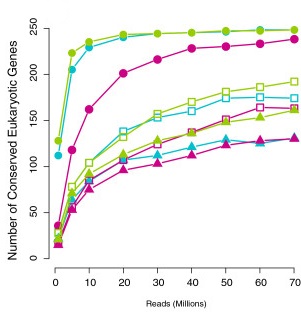
Francis et al. (2013)
Quantification • Counts
- Read counts = gene expression
- Intersection on gene models
- Reads can be quantified on any feature (gene, transcript, exon etc)


Quantification
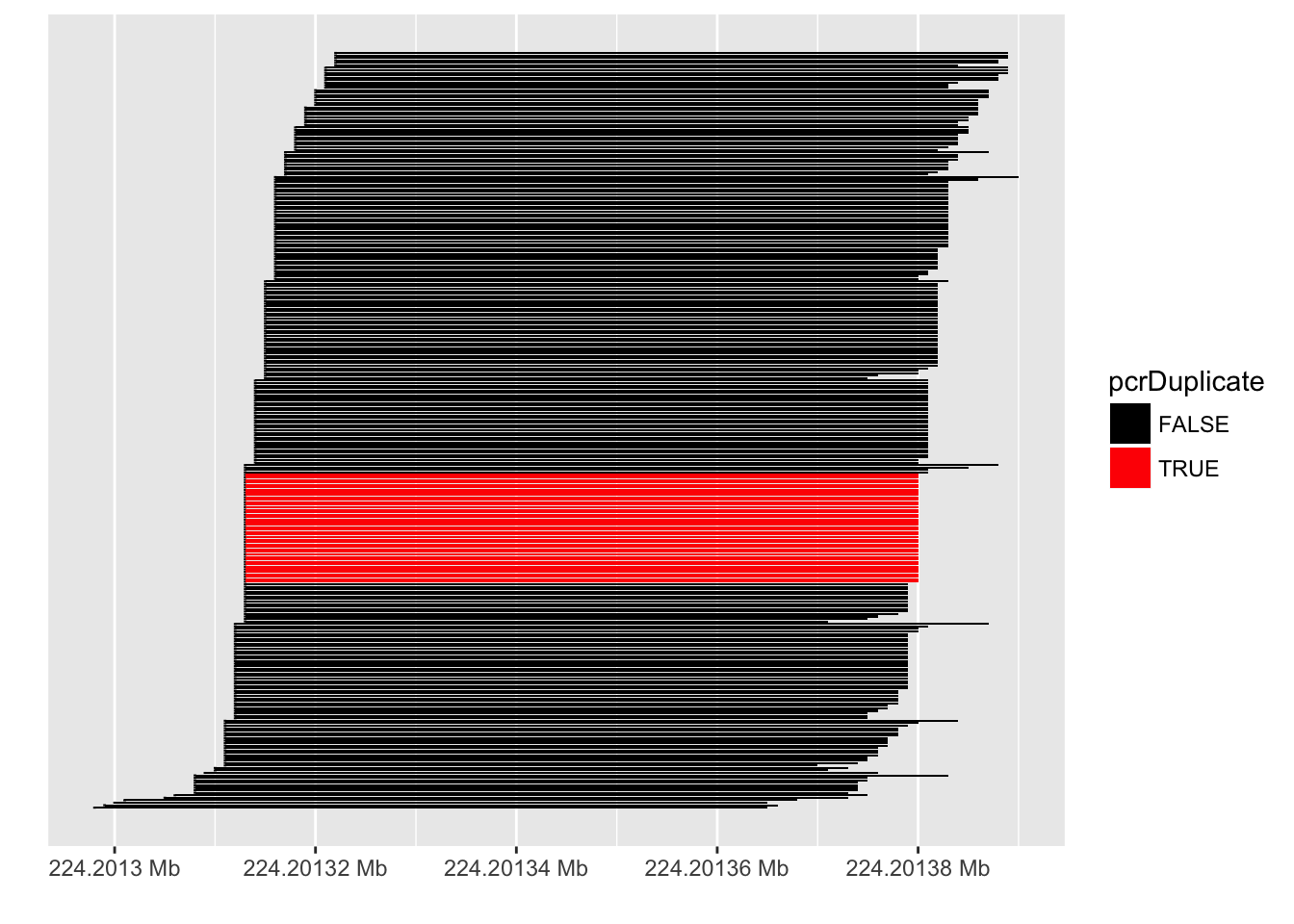
PCR duplicates
- Computational deduplication not recommended Klepikova et al. (2017), Parekh et al. (2016)
- Use PCR-free library-prep kits
- Use UMIs during library-prep Fu et al. (2018)
Multi-mapping
- Added (BEDTools multicov)
- Discard (featureCounts, HTSeq)
- Distribute counts (Cufflinks, featureCounts)
- Rescue
- Probabilistic assignment (Rcount, Cufflinks)
- Prioritise features (Rcount)
- Probabilistic assignment with EM (RSEM)
Quantification QC
ENSG00000000003 140 242 188 143 287 344 438 280 253
ENSG00000000005 0 0 0 0 0 0 0 0 0
ENSG00000000419 69 98 77 55 52 94 116 79 69
ENSG00000000457 56 75 104 79 157 205 183 178 153
ENSG00000000460 33 27 23 19 27 42 69 44 40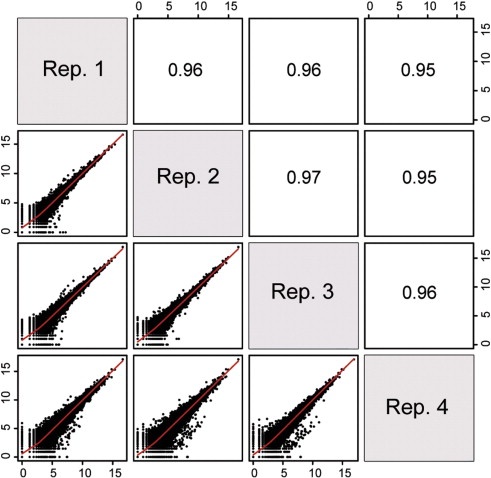
Pairwise correlation between samples must be high (>0.9)
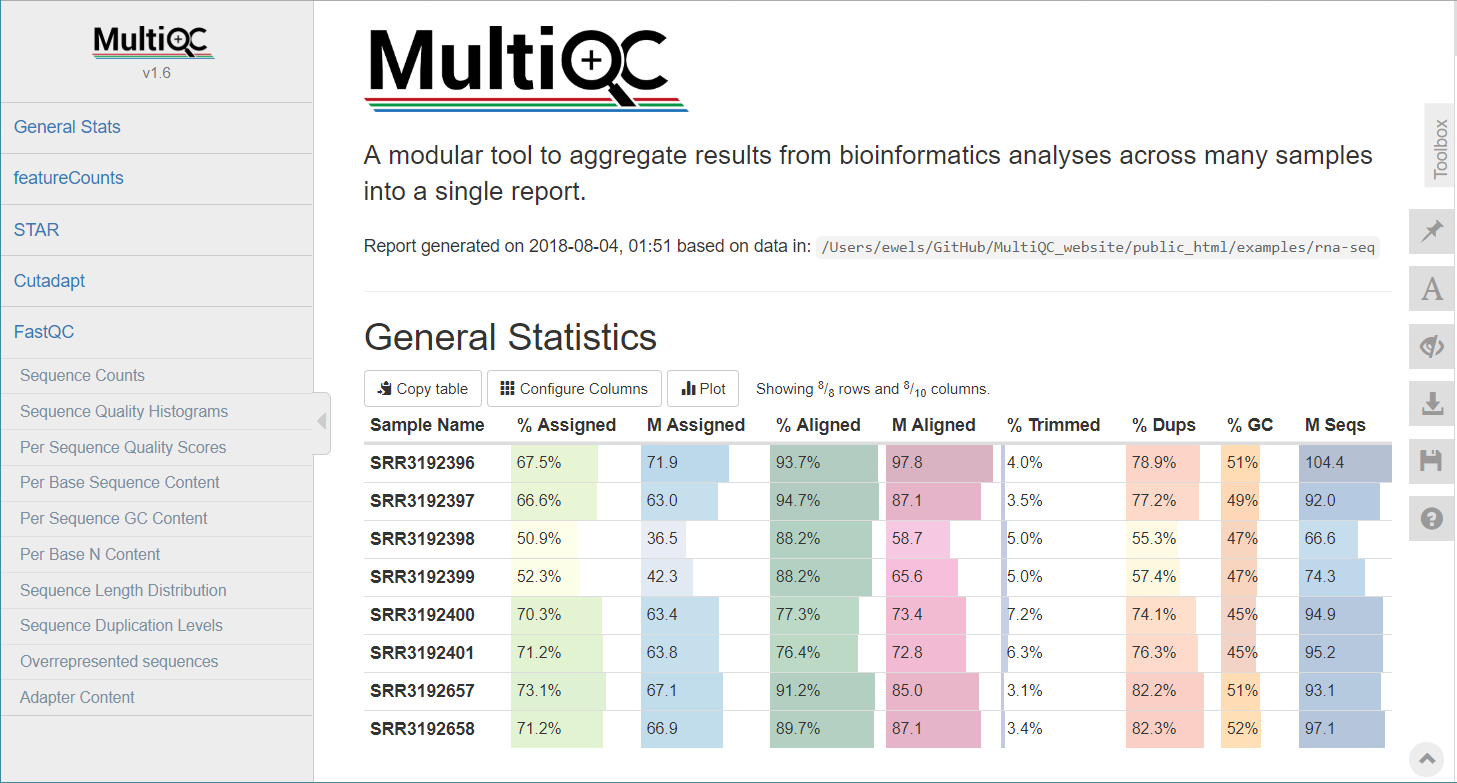
MultiQC
Normalization
- Control for Sequencing depth, compositional bias and more
- Median of Ratios (DESeq2) and TMM (edgeR) perform the best
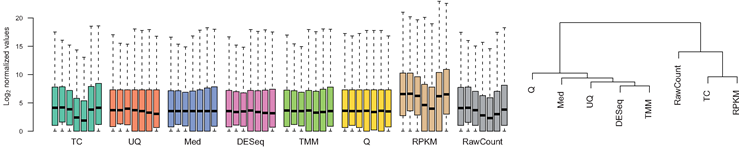
- For DGE using DGE packages, use raw counts
- For clustering, heatmaps etc use VST, VOOM or RLOG
- For own analysis, plots etc, use TPM
- Other solutions: spike-ins/house-keeping genes
Dillies et al. (2013), Evans et al. (2018), Wagner et al. (2012)
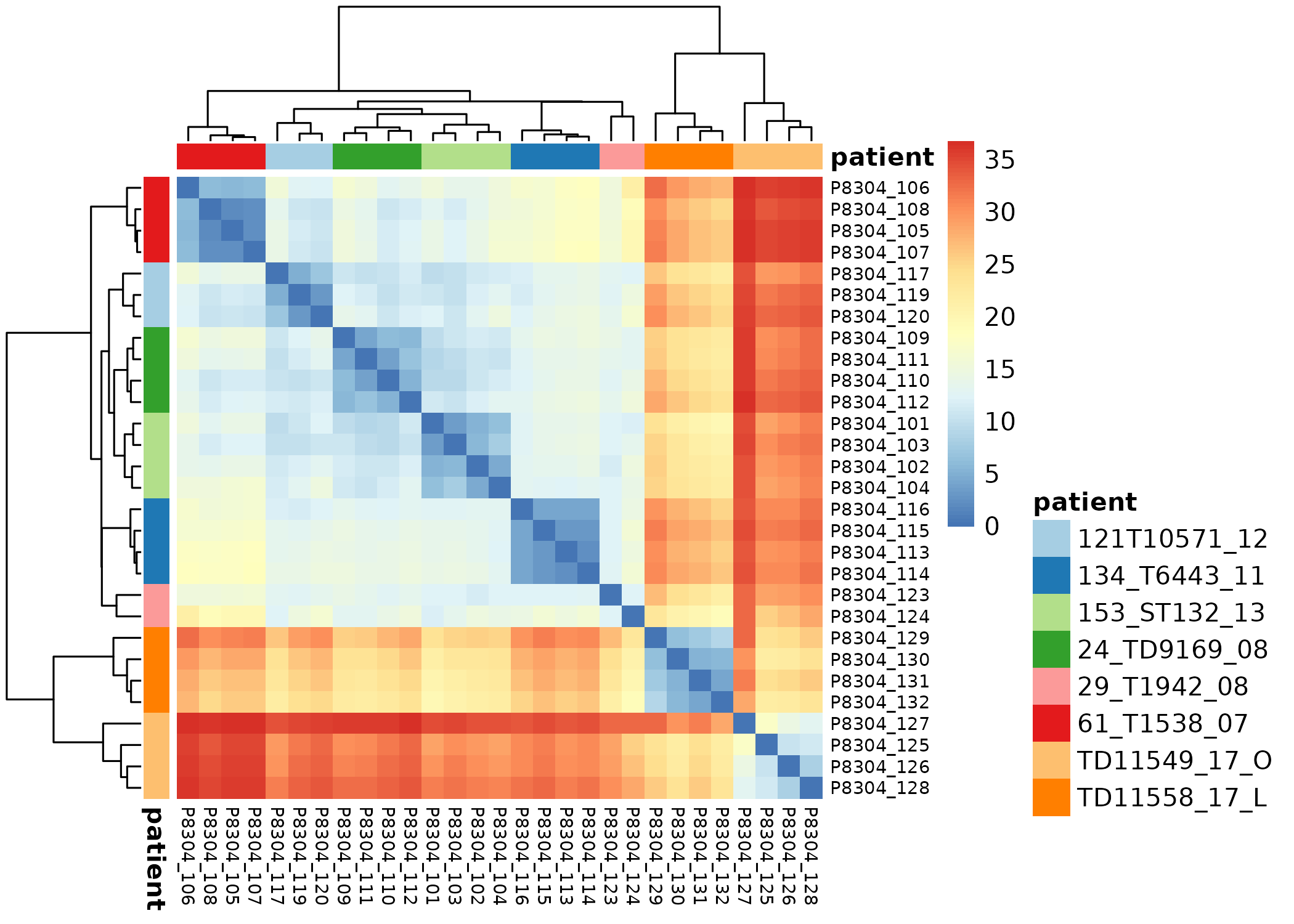
Exploratory
- Transform raw counts to VST, VOOM, RLOG, TPM etc
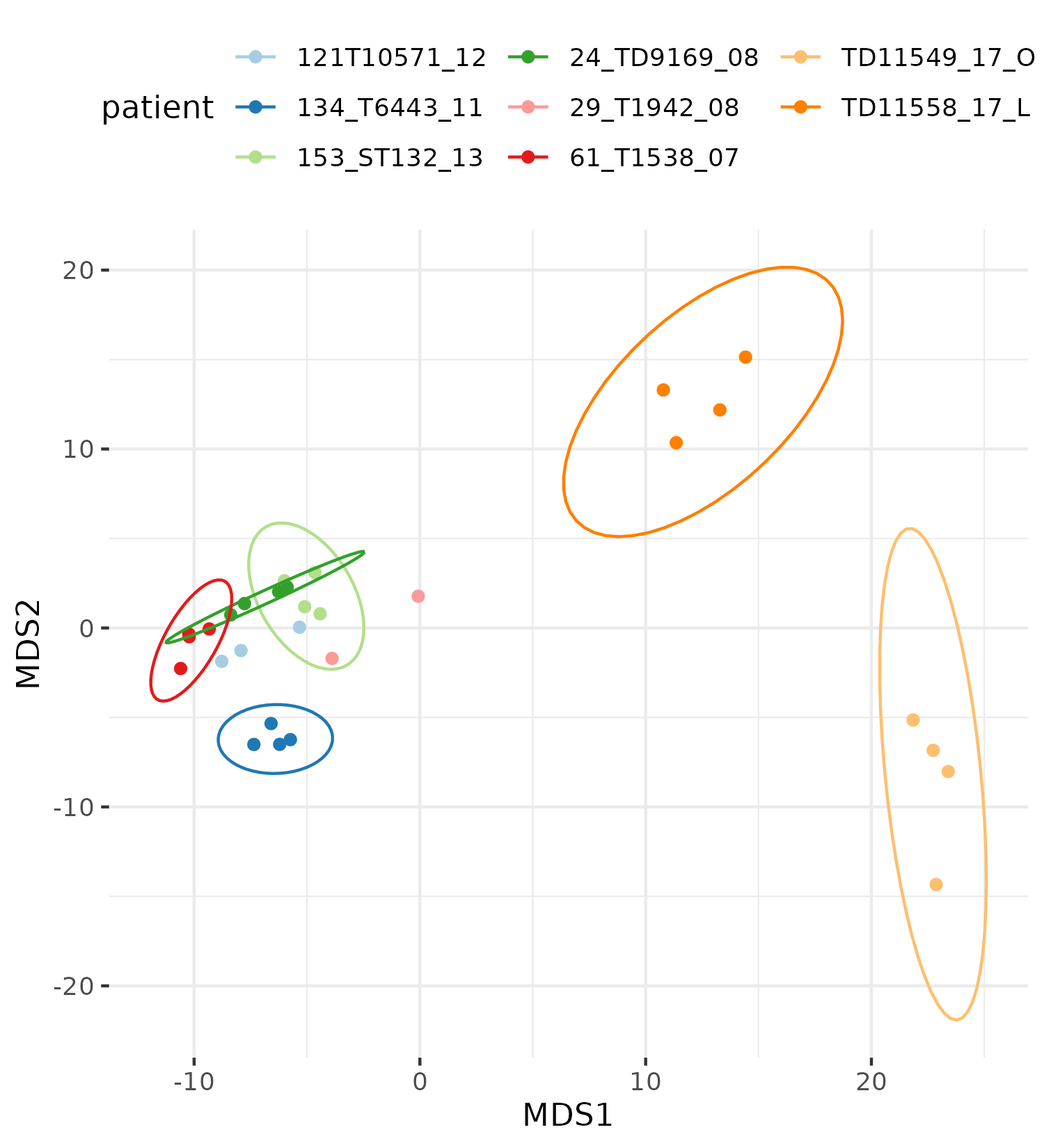
Batch correction
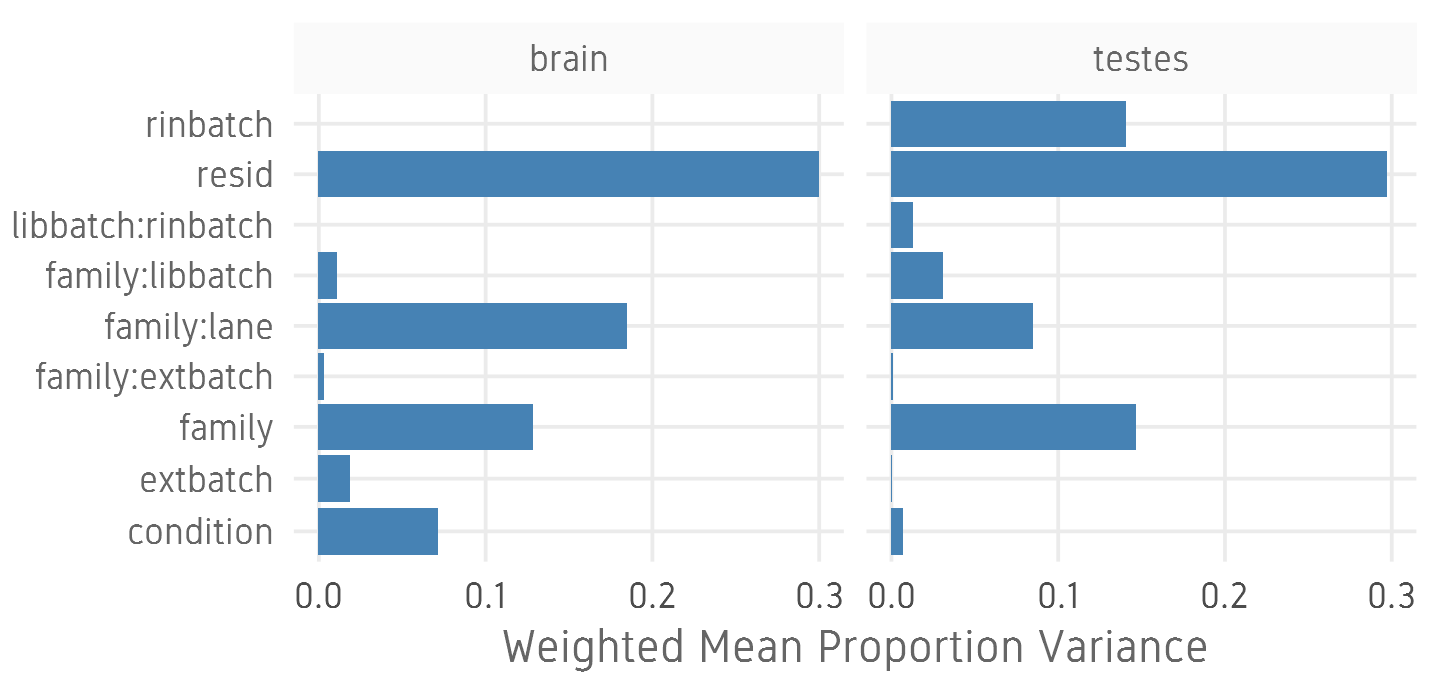
- Estimate variation explained by variables (PVCA)
Differential expression
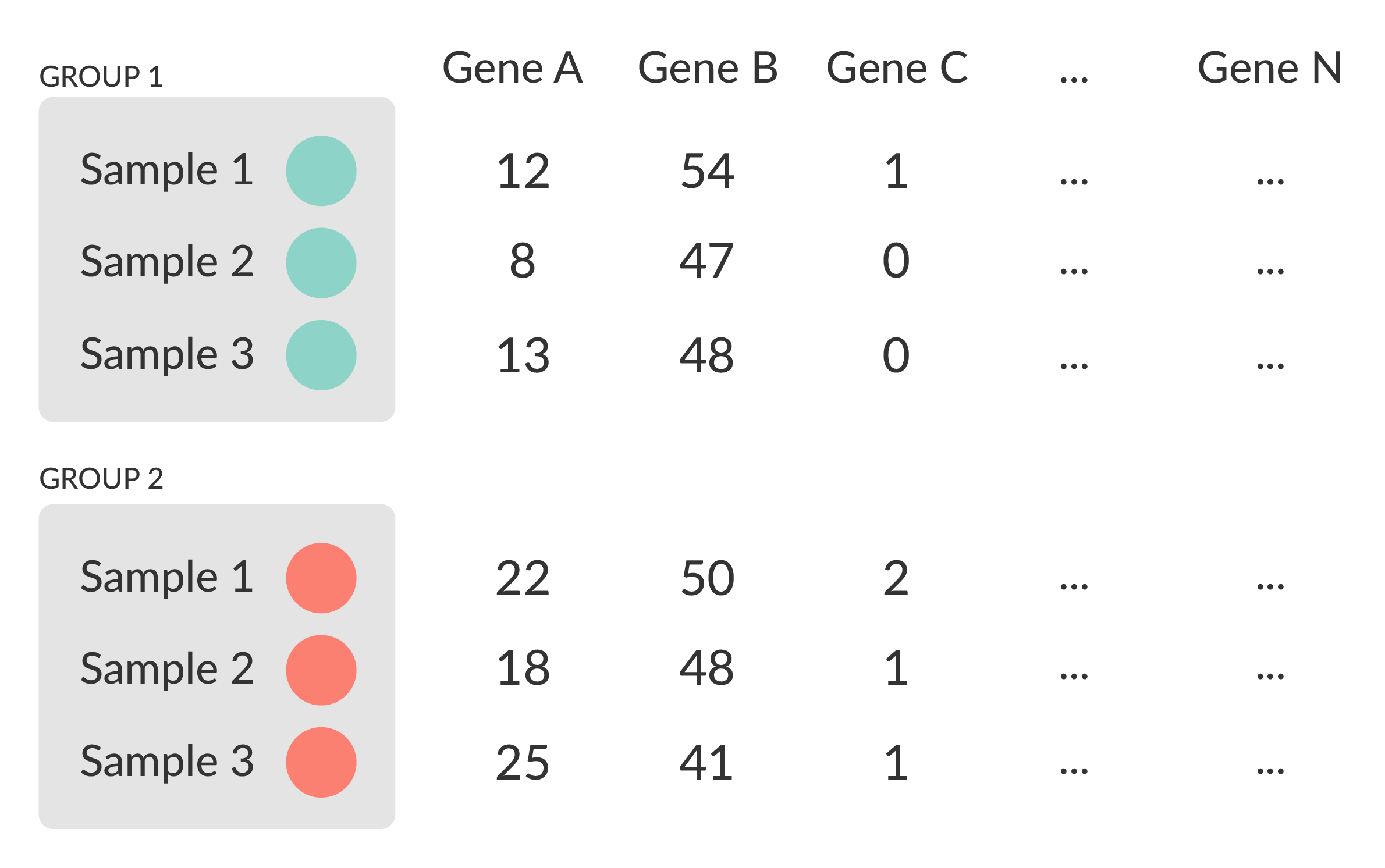
- Univariate testing gene-by-gene
- More descriptive, less predictive
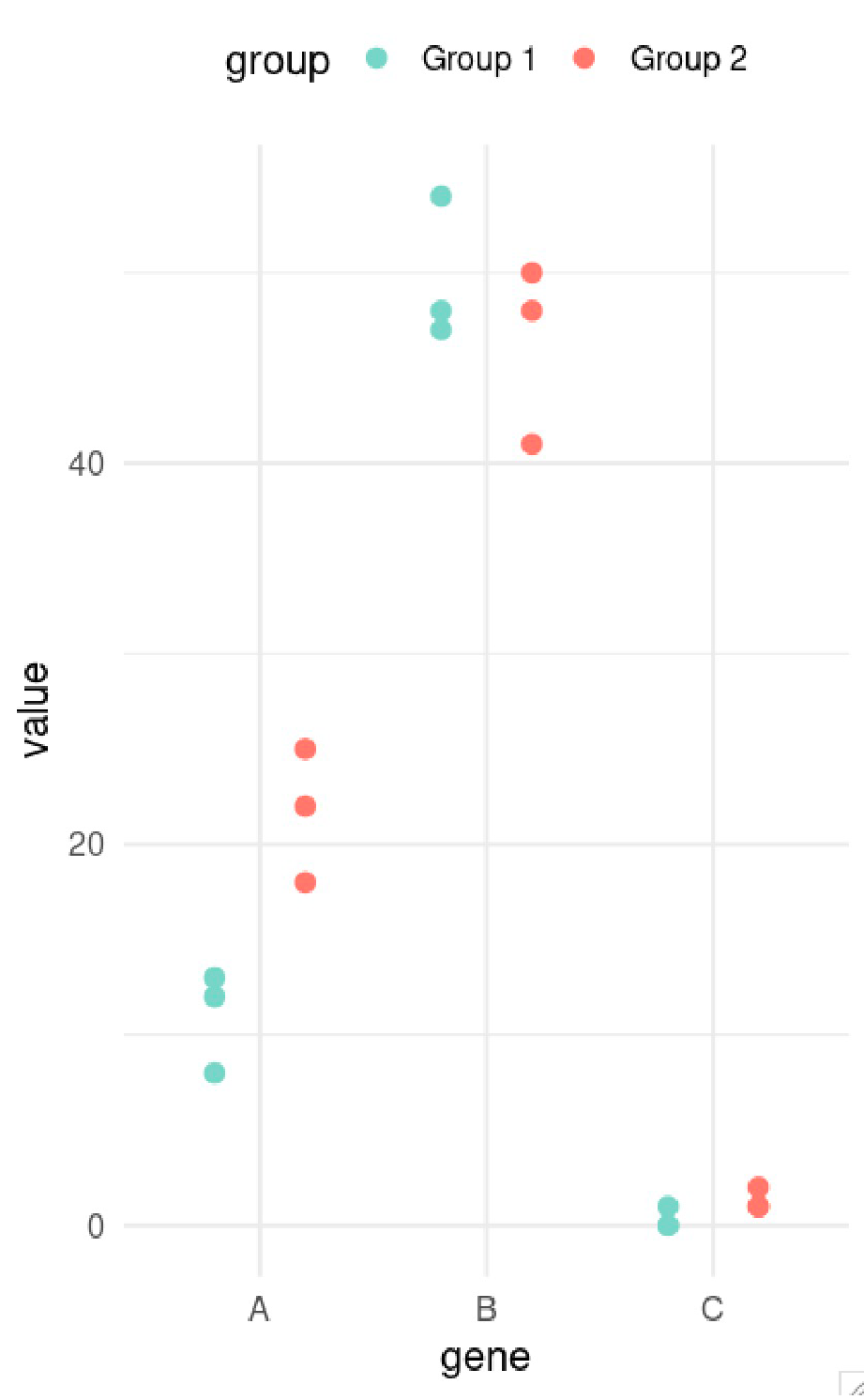
Differential expression
- DESeq2, edgeR (Neg-binom > GLM > Test)
- Limma-Voom (Neg-binom > Voom-transform > LM > Test)
- DESeq2
~age+condition- Estimate size factors
estimateSizeFactors() - Estimate gene-wise dispersion
estimateDispersions() - Fit curve to gene-wise dispersion estimates
- Shrink gene-wise dispersion estimates
- GLM fit for each gene
- Wald test
nbinomWaldTest()
- Estimate size factors
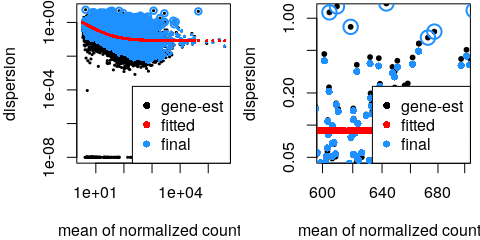
- MA plot
plotMA()
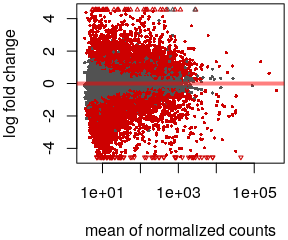
- Volcano plot
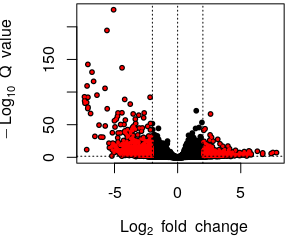
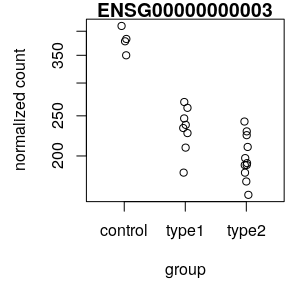
- Normalised counts
plotCounts()

Functional analysis • Gene Ontology
- Gene set analysis (GSA)
- Gene set enrichment analysis (GSEA)
- Gene ontology / Reactome databases
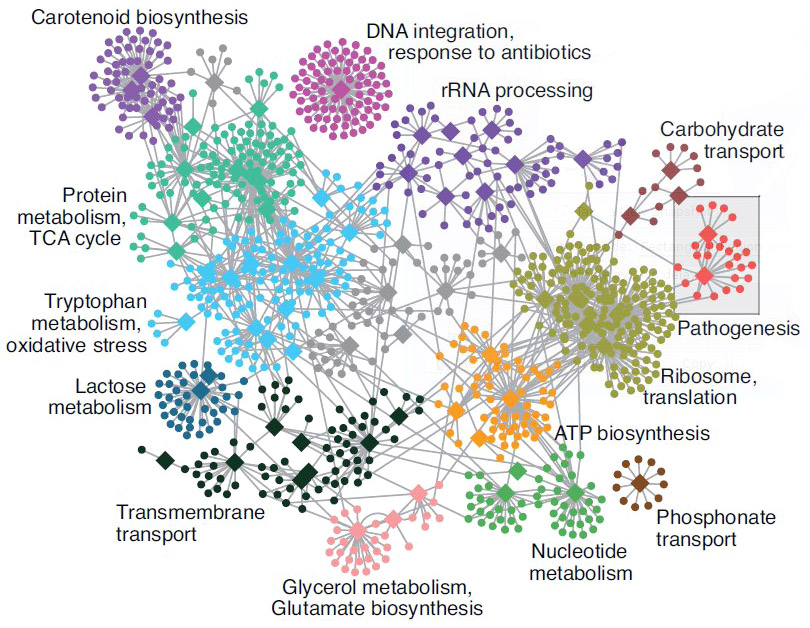
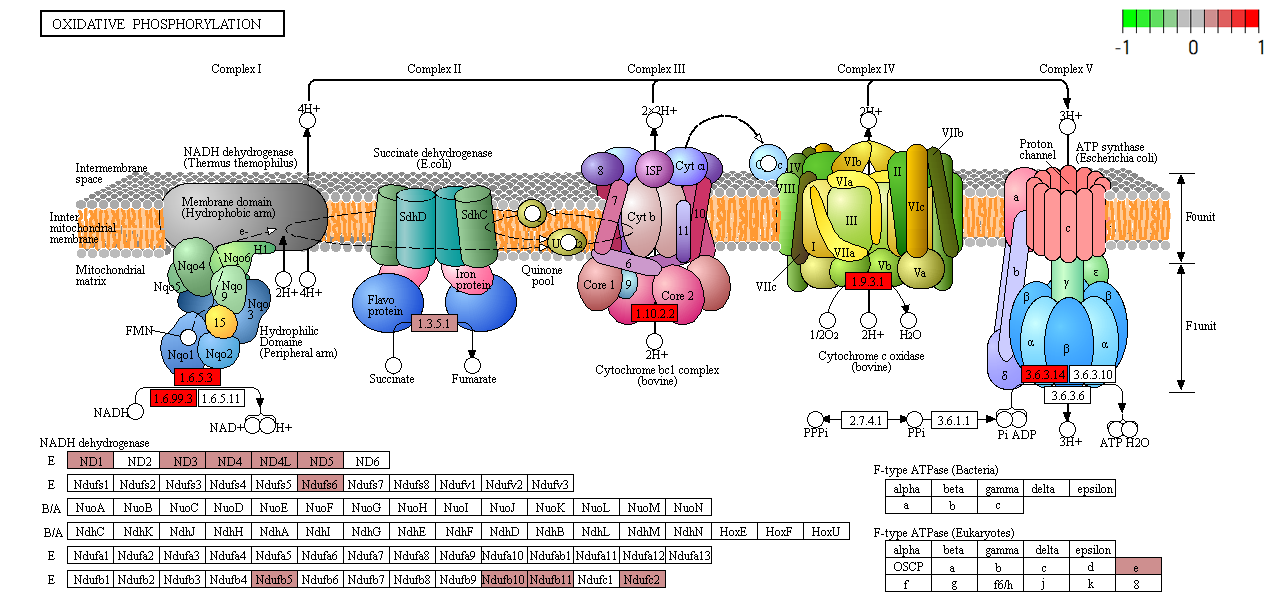
Functional analysis • Kegg
- Pathway analysis (Kegg)
Further learning
- Griffith lab RNA-Seq using HiSat & StringTie tutorial
- HBC Training DGE using DeSeq2 tutorial
- RNA-Seq Blog
- SciLifeLab courses

Hands-On tutorial
- Course data directory
/sw/courses/ngsintro/rnaseq/dardel
dardel/
├── bonus
│ ├── assembly
│ ├── exon
│ ├── funannot
│ └── plots
├── main
│ ├── 1_raw
│ ├── 2_fastqc
│ ├── 3_mapping
│ ├── 4_qorts
│ ├── 4_qualimap
│ ├── 5_dge
│ ├── 6_multiqc
│ ├── reference
│ │ └── mouse_chr19_hisat2
│ └── scripts
├── main_full
│ └── nextflow
├── r
└── README.md- Your work directory
~/ngsintro/rnaseq/
rnaseq/
├── 1_raw
├── 2_fastqc
├── 3_mapping
├── 4_picard
├── 4_qualimap
├── 5_dge
├── 6_multiqc
├── funannot
├── plots
├── reference
└── scripts
