Functions
Paulo Czarnewski
2019-10-24
[1] “/Users/paulo.barenco/Box/repos/sauron/docs/documentation_files”
Introduction
Here we give a deep understanting of the functions and parameters being used in the workflow.
Running with SLURM
You can safely ignore these lines in case you are using the pipeline on a single computer.
The first lines provides a convenient way to run the workflow using the SLURM queueing system on a High Performance Computing environment.
#! /bin/bash -l
#SBATCH -A projID
#SBATCH -p core
#SBATCH -n 8
#SBATCH -J Proj
#SBATCH -t 10:00:00
#SBATCH --mail-user username@email.com
#SBATCH --mail-type=ENDInitializing Sauron.v1 conda environment
You can safely ignore these lines.
The following lines allows the pipeline to find the main project folder (i.e. where the run_workflow.sh is located).
The first will dictate where your data and scripts are. Because of this feature, one can simply define the path to your project as $main. The second will load the script to initiallize the Sauron.v1 conda environment with all necessary packages for the analysis. If you don’t already have the environment, it will create it for you.
main="$( cd "$( dirname "${BASH_SOURCE[0]}" )" >/dev/null 2>&1 && pwd )"
source $main/initialize.sh 2>&1 | tee $main/log/'initialize_log.txt'Global parameters
In these lines you can define global parameters to be user across your analysis. They should either come from the metadata file for each dataset you will provide (see below), or from the pipeline itself (see below).
These names are essentially column names in your metadata inside the Seurat Object.
By default in the Quality Control step (see below), several metrics are autmatically put into the seurat object, and those can be used for plotting here. Some of them are: nCounts_RNA, nFeature_RNA , percent_rps , percent_rpl , percent_ribo , S.Score , G2M.Score.
var_to_plot='VARIABLE_COLUMNS_FROM_METADATA_TABLE,percent_mito,percent_ribo'
var_to_regress='nFeature_RNA,percent_mito,S.Score,G2M.Score'Load datasets
Rscript $script_path/00_load_data.R \
--input_path $main/'data' \
--dataset_metadata_path $main/'data/metadata.csv' \
--assay 'RNA' \
--output_path $main/'analysis/1_qc' \
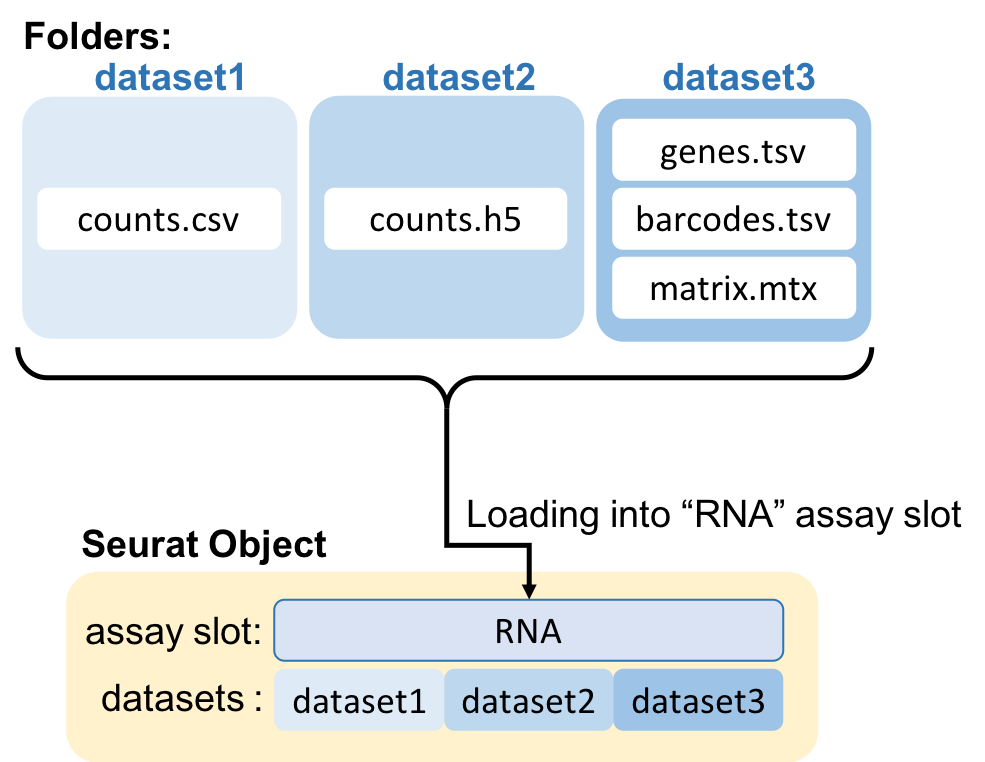
2>&1 | tee $main/log/'00_load_data_log.txt'input_pathThe PATH to the FOLDER containing your datasets (as folders). Each folder inside this PATH is a dataset.
dataset_metadata_pathThe PATH to the metadata FILE for each library (The first column should be named SampleID)
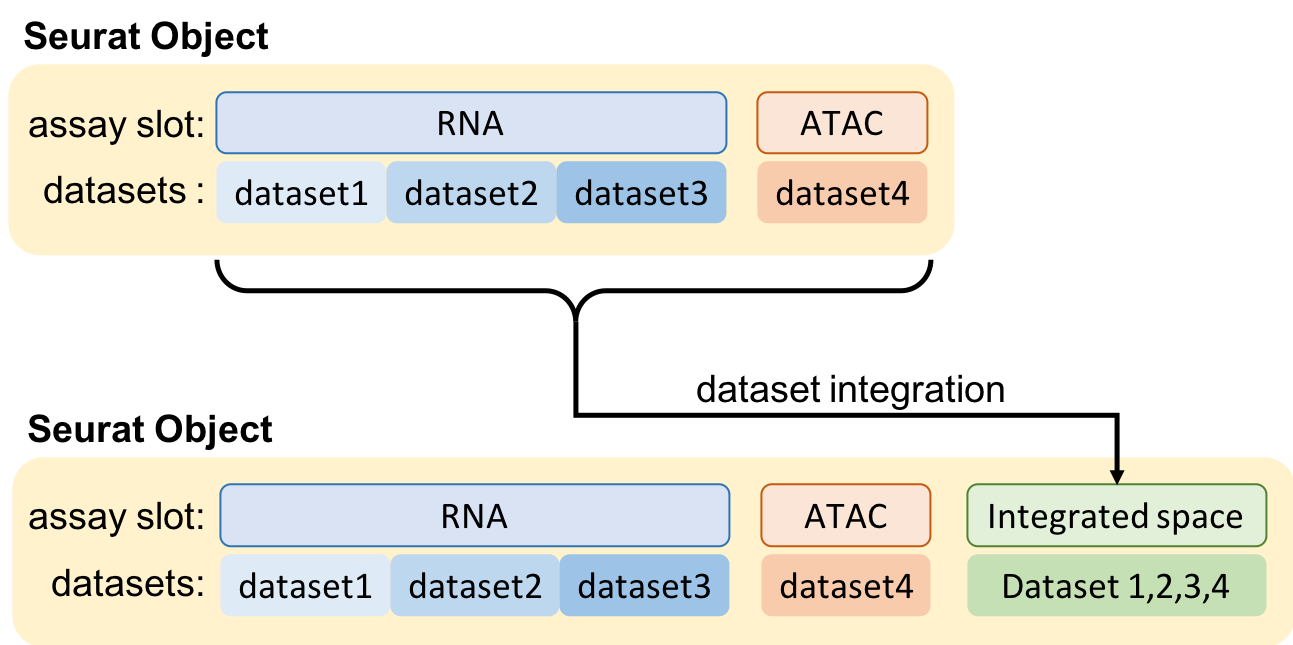
assayThe ASSAY slot in the Seurat object to load the data. Default is “RNA”. You can add any name that describes the type of data in that assay: “CITE”, “ATAC”, “PROTEIN”, etc.
output_pathThe PATH to the FOLDER to output your Seurat object.
To load datasets, first place them in the data folder: create a folder for each individual dataset, and within each folder, place your dataset (see figure below). This allows a well organized rawdata folder for the analysis.
Besides the rawdata, you can add a metadata .csv file, which can contain information about each specific dataset. Each line is a dataset. Each column is a metadata variable. All those values will be parsed to the Seurat object on the loading.
The loading function will also automatically take care of finding the union of all genes across datasets and adding zeros where the genes are not expressed.
Run Quality Control
Rscript $script_path/01_qc_filter.R \
--Seurat_object_path $main/'analysis/1_qc/raw_seurat_object.rds' \
--columns_metadata $var_to_plot \
--species_use 'mmusculus' \
--remove_non_coding 'True' \
--plot_gene_family 'RPS,RPL,mito,HB' \
--remove_gene_family 'mito' \
--min_gene_count '5' \
--min_gene_per_cell '200' \
--assay 'RNA' \
--output_path $main/analysis/1_qc \
2>&1 | tee $main/log/'01_QC_log.txt'Seurat_object_pathThe PATH to the Seurat Object containing your datasets and metadata.
columns_metadataThe COLUMN NAMES of the metadata matrix to load into the objects. They will be treated as factors variables (not continuous).
species_useSpecies from the sample for cell cycle scoring.
remove_non_codingRemoves all non-coding and pseudogenes from the data. Default is ‘True’.
plot_gene_familyGene families to calculate and add to the metadata, comma separated. You can place them as patterns of genes or gene families. Some examples are: “RPS” (ribosomal), “RPL” (ribosomal), “MT-” (mitocondria). Upper and Lower case are ignored. Default is “Rps,Rpl,mt-,Hb”.
remove_gene_familyGene families to remove from the count matrix after QC, comma separated. They should start with the pattern.Upper and Lower case are ignored. Default is “mt-”.
min_gene_countMinimun number of cells needed to consider a gene as expressed. Default is 5.
min_gene_per_cellMinimun number of genes in a cell needed to consider a cell as good quality. Default is 200.
assayThe ASSAY slot in the Seurat object to load the data. Default is “RNA”. You can add any name that describes the type of data in that assay: “CITE”, “ATAC”, “PROTEIN”, etc.
output_pathThe PATH to the FOLDER to output your Seurat object.
Integrating datasets
Rscript $script_path/02_integrate.R \
--Seurat_object_path $main/'analysis/1_qc/filt_seurat_object.rds' \
--columns_metadata $var_to_plot \
--regress $var_to_regress \
--var_genes 'seurat' \
--integration_method 'mnn,sample_id' \
--cluster_use 'NONE' \
--assay 'RNA' \
--output_path $main/'analysis/2_clustering' \
2>&1 | tee $main/log/'02_integrate_log.txt'Seurat_object_pathThe PATH to the Seurat Object containing your datasets and metadata.
columns_metadataThe COLUMN NAMES of the metadata matrix to load into the objects. They will be treated as factors variables (not continuous).
regressVariables of the metadata to be regressed out using GLM.
var_genesWhether use ‘Seurat’ or the ‘Scran’ method for variable genes identification. An additional value can be placed after a comma to define the level of dispersion wanted for variable gene selection. ‘Seurat,2’ will use the threshold 2 for gene dispersions. Defult is ‘Seurat,1.5’. For Scran, the user should inpup the level of biological variance ‘Scran,0.2’. An additional blocking parameter (a column from the metadata) can ba supplied to ‘Scran’ method block variation comming from uninteresting factors, which can be parsed as ‘Scran,0.2,Batch’.
integration_methodIntegration method to be used. ‘CCA’, MNN’, ‘Scale’ and ‘Combat’ are available at the moment. The batches (column names in the metadata matrix) to be removed should be provided as arguments comma separated. E.g.: ‘Combat,sampling_day’. For MNN, an additional integer parameter is supplied as the k-nearest neighbour.
cluster_useThe cluster to be used for analysis parsed as “METADATA_COLUMN_NAME,factor1,factor2”. This is usefull if you want to subset your data and perform this step in only part of the dataset (a cell cluster, or a particular metadata parameter). Example: “SNN.0.4,1,2,3,4”, will get clusters 1-4 from the the metadata column “SNN.0.4”.
assayThe ASSAY slot in the Seurat object to load the data. Default is “RNA”. You can add any name that describes the type of data in that assay: “CITE”, “ATAC”, “PROTEIN”, etc.
output_pathThe PATH to the FOLDER to output your Seurat object.
Dim. reduction and Clustering
Rscript $script_path/03_dr_and_cluster.R \
--Seurat_object_path $main/'analysis/2_clustering/seurat_object.rds' \
--columns_metadata $var_to_plot \
--regress $var_to_regress \
--PCs_use 'var,1' \
--var_genes 'seurat' \
--dim_reduct_use 'umap' \
--cluster_use 'none' \
--cluster_method 'HC,kmeans' \
--assay 'RNA' \
--output_path $main/'analysis/2_clustering' \
2>&1 | tee $main/log/'03_dr_and_cluster_log.txt'Seurat_object_pathcolumns_metadataregressPCs_usevar_genesdim_reduct_usecluster_usecluster_methodassayoutput_path
Clustering correlation analysis
Rscript $script_path/'05_cluster_correlation.R' \
--Seurat_object_path $main/'analysis/2_clustering/seurat_object.rds' \
--clustering_use 'HC_100' \
--exclude_cluster 'NONE' \
--merge_cluster '0.95,0.9,0.85,0.8,0.75,0.7' \
--output_path $main/'analysis/2_clustering/cluster_correlations' \
2>&1 | tee $main/'log/4_clust_corr.txt'Differential gene expression
Rscript $script_path/04_diff_gene_expr.R \
--Seurat_object_path $main/'analysis/2_clustering/seurat_object.rds' \
--clustering_use 'HC_12' \
--metadata_use 'tech' \
--exclude_cluster 'NONE' \
--assay 'RNA' \
--o $main/'analysis/3_diff_expr' \
2>&1 | tee $main/'log/04_diff_expr_log.txt'Trajectory inference analysis
Also refered as “pseudotime”.
Rscript $script_path/07_trajectory.R \
--Seurat_object_path $main/'analysis/2_clustering/seurat_object.rds' \
--metadata_use '' \
--reduction_use '' \
--reduction_visualize '' \
--method_use '' \
--no_traj_components '' \
--no_of_paths '' \
--start_cluster '' \
--assay '' \
--output_path '' \
2>&1 | tee $main/'log/07_trajectory_log.txt'Cell type classification
Rscript $script_path/cell_type_prdiction.R \
--Seurat_object_path $main/'analysis/2_clustering/seurat_object.rds' \
--marker_lists $main/'marer_list.csv' \
--cluster_use 'snn_0.4' \
--assay 'RNA' \
--output_path $main/'analysis/2_clustering/' \
2>&1 | tee $main/'log/cell_type_prediction_log.txt'Plotting custom gene lists
Rscript $script_path/plot_gene_list.R \
--Seurat_object_path $main/'analysis/2_clustering/seurat_object.rds' \
--gene_list $main/'my_genes_of_interest.csv' \
--match_type 'exact' \
--clustering_use 'snn_0.4' \
--assay 'RNA' \
--output_path $main/'analysis/2_clustering/' \
2>&1 | tee $main/'log/plot_gene_list_log.txt'Ligand-Receptor interaction
Rscript $script_path/06_lig_rec_interactome.R \
--objects_paths $main/'analysis/2_clustering/seurat_object.rds' \
--object_names 'all_cells' \
--object_clusters 'HC_12,1,2,3,4' \
--lig_recp_database 'DEFAULT' \
--ligand_objects 'all_cells' \
--receptor_objects 'all_cells' \
--species_use 'hsapiens' \
--metadata_ligands 'tech' \
--metadata_receptor 'tech' \
--filter_thresholds '0.1,0.1,3' \
--output_path $main/'analysis/5_Lig_Rec_interaction' \
--assay 'RNA' \
2>&1 | tee $main/'log/06_Interactome_EPI_log.txt'VDJ analysis