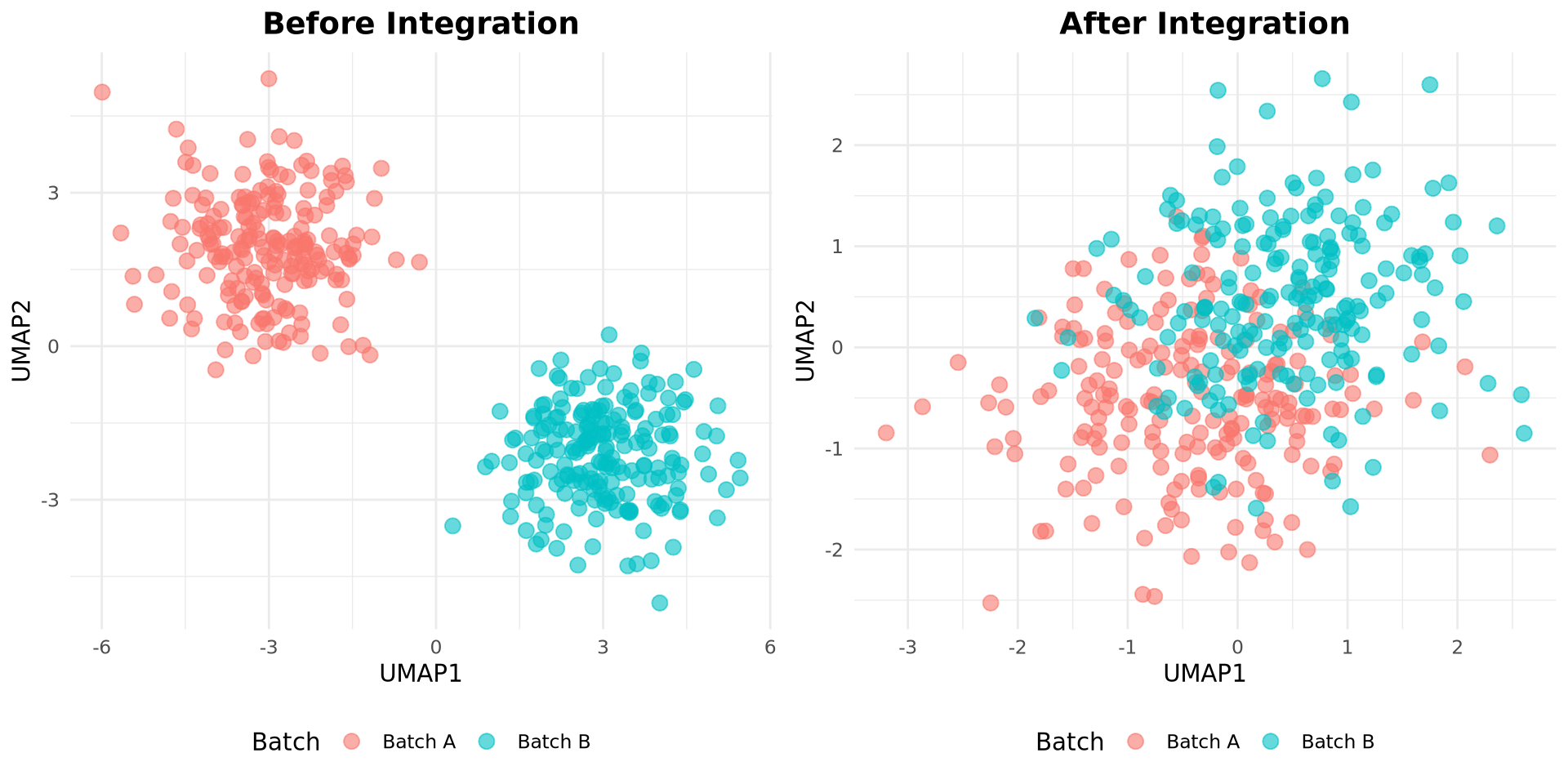
scRNA-seq Integration & Batch correction
Multi-sample/batch harmonization
14-Apr-2026
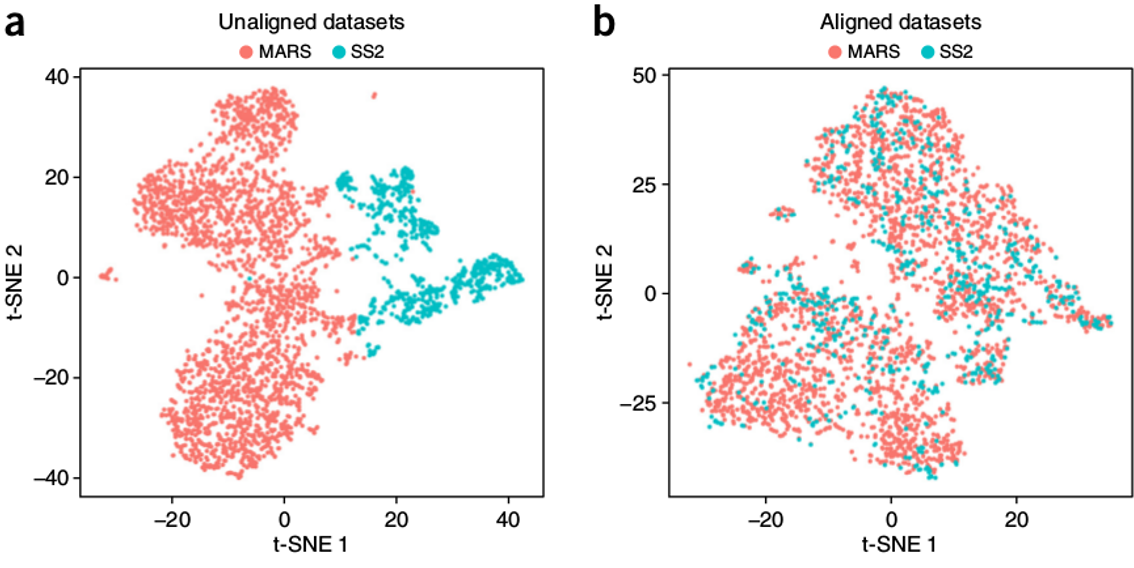
Closer look
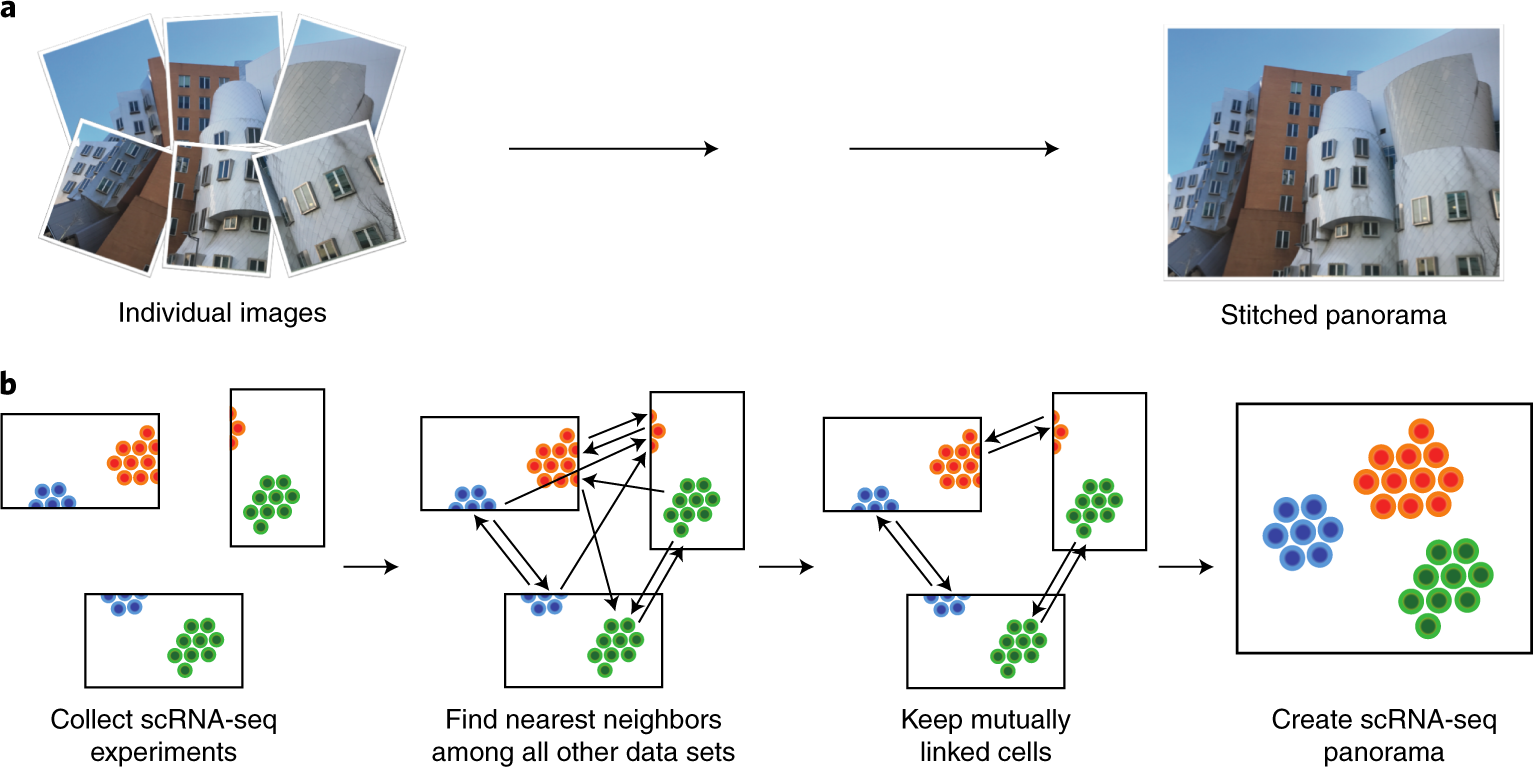
- Panel “a”: Same type of data (scRNAseq)
- Integration & batch-correction (BC)
- 3 samples same run: Without BC
- 3 samples 2 batches: With BC
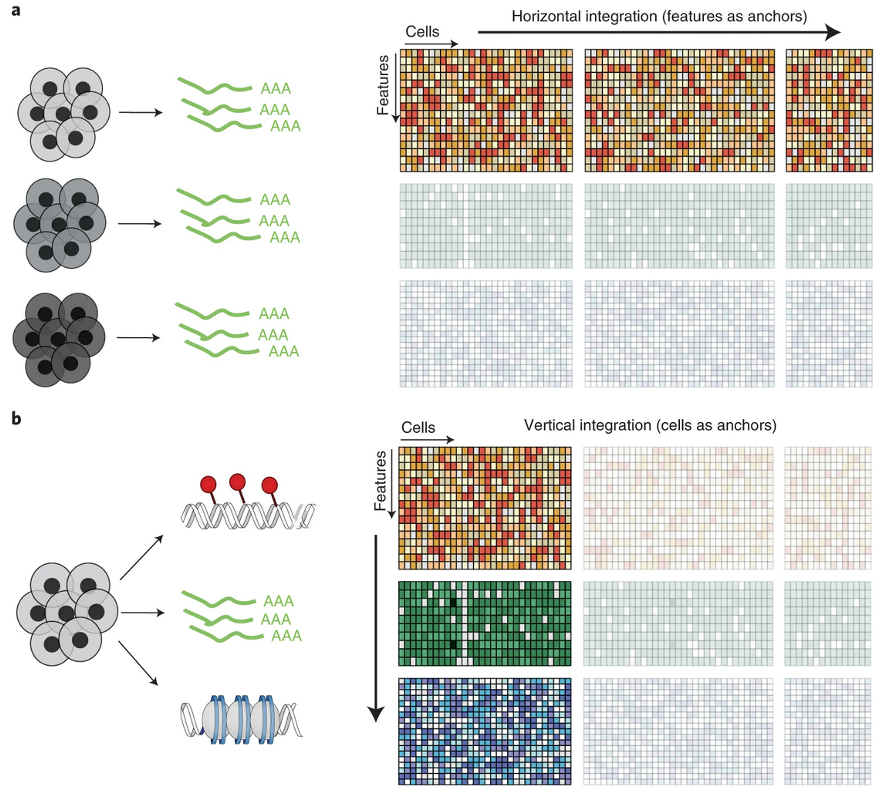
- Panel “b”: Multi-omics Integration
- Same samples, multiple platforms
- Example: RNAseq + ATACseq
- Beyond scope here
- Good to know and not to confuse
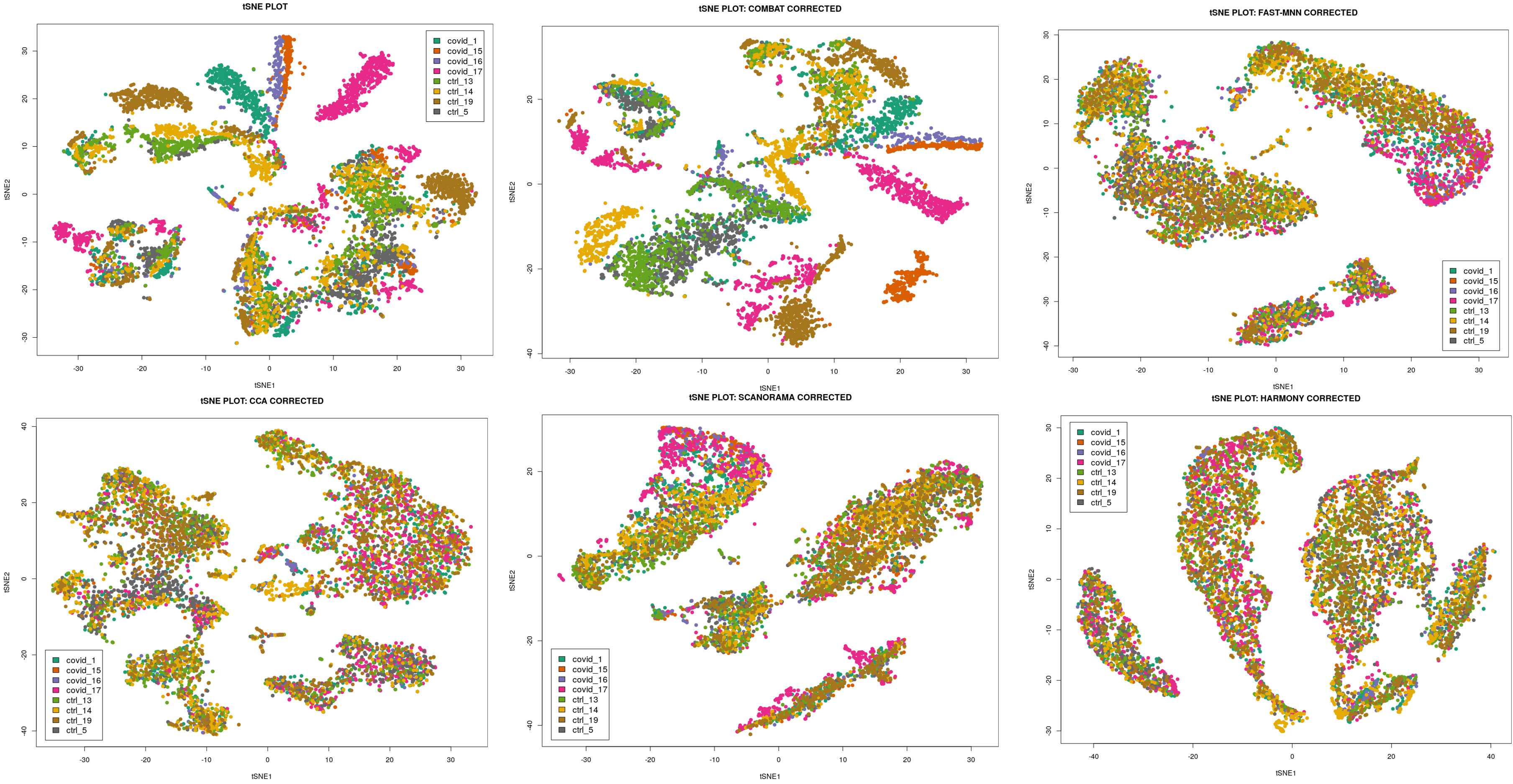
CCA: Canonical Correlation Analysis
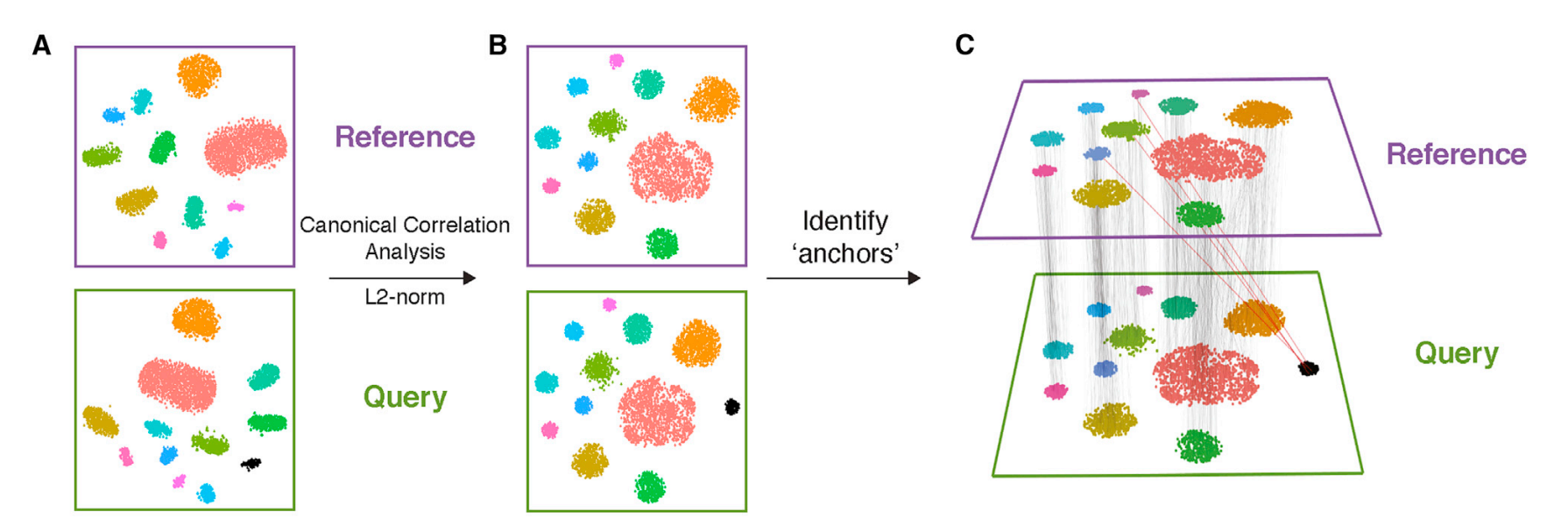
- Finds correlated features between batches
- Creates canonical variates capturing shared variation
- Projects cells into aligned latent space
- Linear approach - computationally efficient for large datasets
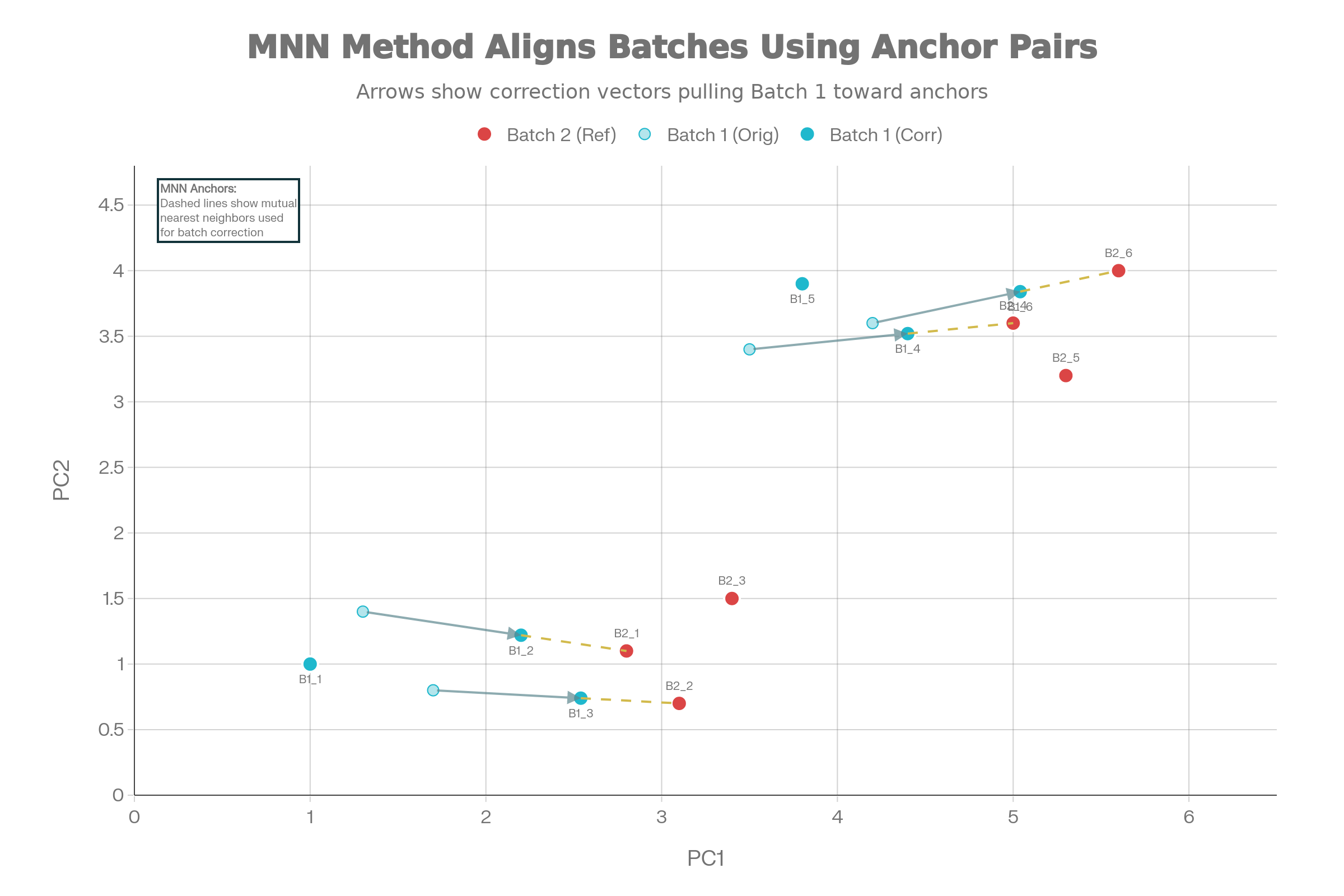
Visualizing MNN Correction
Harmony: Iterative PC Correction
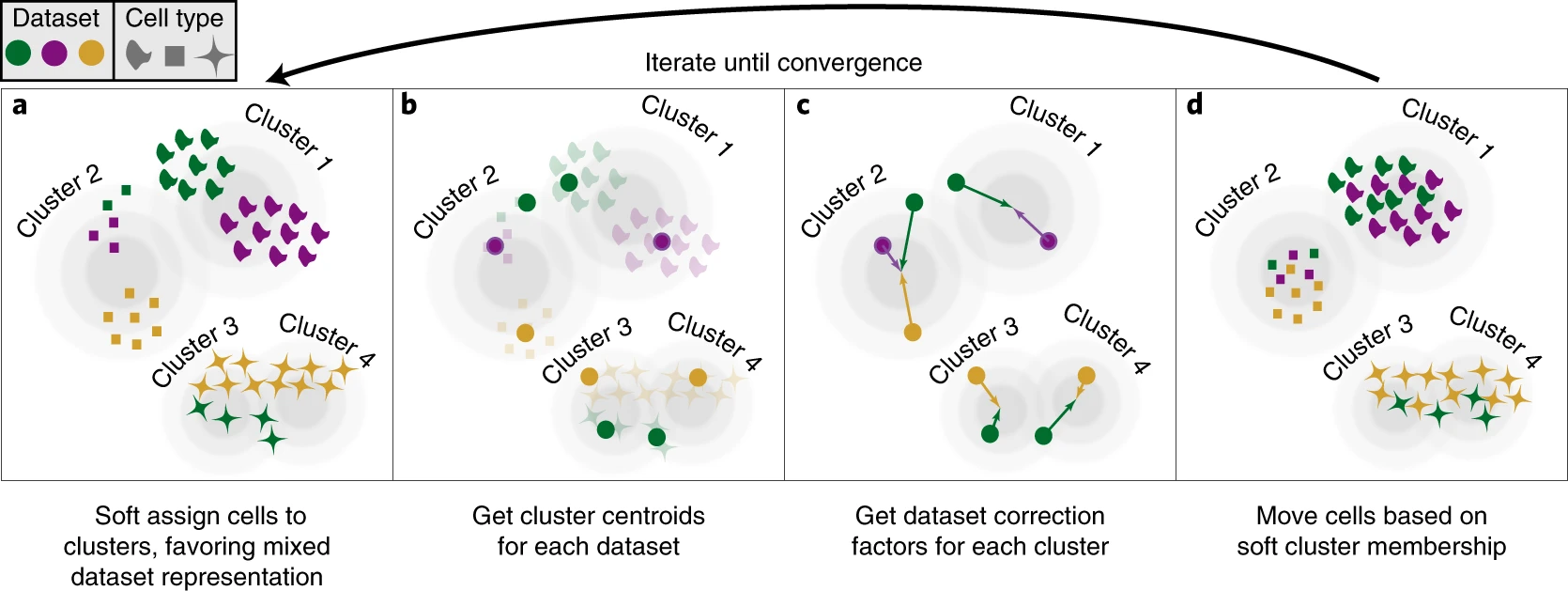
Harmony iteratively adjusts cell embeddings so that each cluster contains a balanced mix of batches
Scanorama: Manifold Alignment + SVD
- Builds manifold alignment between pairs of datasets
- Uses Singular Value Decomposition (SVD) for efficient merging
- Iteratively corrects and merges multiple datasets
Conos: Graph-Based Alignment
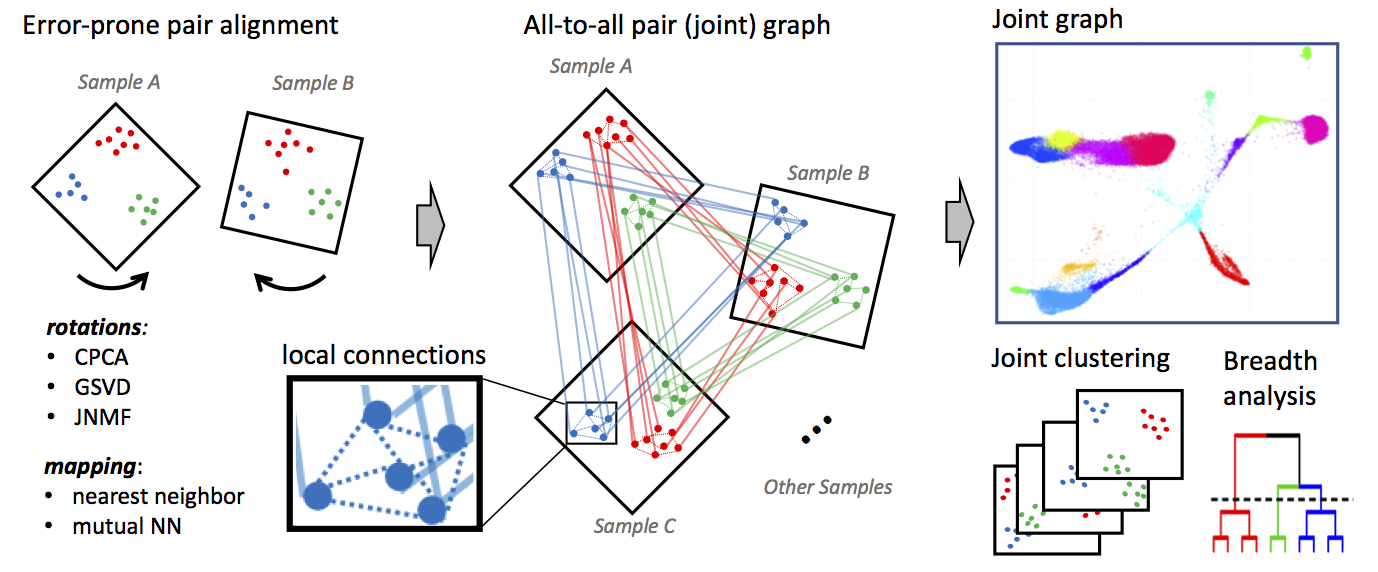
- Builds separate k-NN graphs for each sample
- Constructs joint graph by adding cross-sample edges between similar cells
- Preserves per-sample structure while enabling global analysis
- Particularly useful for many-sample studies with distinct biological states
Comparison
Resources & Further reading
- Luecken, M.D., Büttner, M., Chaichoompu, K. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat Methods 19, 41–50 (2022). https://doi.org/10.1038/s41592-021-01336-8
- Korsunsky, I., Millard, N., Fan, J. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). https://doi.org/10.1038/s41592-019-0619-0
- Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat Biotechnol 37, 685–691 (2019). https://doi.org/10.1038/s41587-019-0113-3
- Argelaguet, R., Velten, B., Arnol,D, S. et al. Multi-omics factor analysis—a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol 14, e8124 (2018). https://doi.org/10.15252/msb.20178124
- Barkas N., Petukhov V., Nikolaeva D., Lozinsky Y., Demharter S., Khodosevich K., & Kharchenko P.V. Joint analysis of heterogeneous single-cell RNA-seq dataset collections. Nature Methods, (2019). https://doi.org/10.1038/s41592-019-0466-z

