@HD VN:1.6 SO:coordinate
@SQ SN:LG4 LN:100000
@RG ID:SRR9309790 SM:PUN-Y-INJ PL:ILLUMINA
@PG ID:bwa PN:bwa VN:0.7.19-r1273 CL:bwa mem -R @RG\tID:SRR9309790\tSM:PUN-Y-INJ\tPL:ILLUMINA -p -t 30 -M tiny/ref/M_aurantiacus_v1_splitline_ordered.fasta -Read mapping
Mapping reads to a reference sequence
Sequence alignment maps reads to a reference
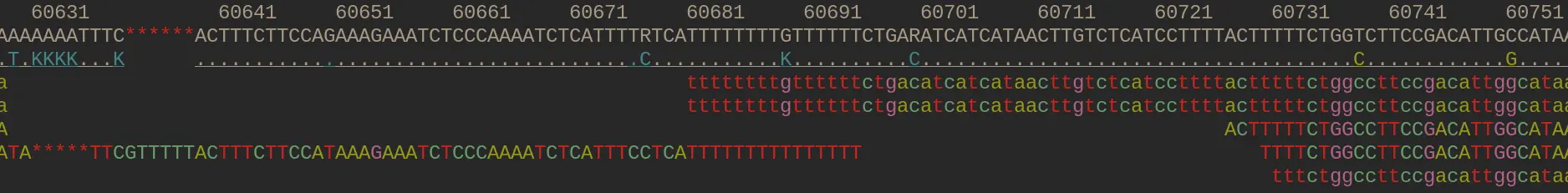
. characters is the consensus sequence. Bases are colored by nucleotide. Letter case indicates forward (upper-case) or reverse (lower-case) alignment. * is placeholder for deleted base.
Aim of sequence alignment (read mapping) is to determine source in reference sequence. Some commonly used read mappers for resequencing are
- BWA, BWA-MEM (Li, 2013)
- Novoalign (https://www.novocraft.com/)
- Minimap2 (Li, 2018)
For a recent comprehensive comparison see Donato et al. (2021)
Bibliography
Donato, L., Scimone, C., Rinaldi, C., D’Angelo, R., & Sidoti, A. (2021). New evaluation methods of read mapping by 17 aligners on simulated and empirical NGS data: An updated comparison of DNA- and RNA-Seq data from Illumina and Ion Torrent technologies. Neural Computing and Applications, 33(22), 15669–15692. https://doi.org/10.1007/s00521-021-06188-z
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-Bio]. https://arxiv.org/abs/1303.3997
Li, H. (2018). Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics, 34(18), 3094–3100. https://doi.org/10.1093/bioinformatics/bty191