
DNA sequencing data
DNA sequencing approaches and data
Sequencing technologies

![]()
Illumina NovaSeq 600
Scale up and down with a tunable output of up to 6 Tb and 20B single reads in < 2 days.
Up to 2X250 bp read length. Price example: 8,000 SEK total for resequencing 3Gbp genome to 30X
PacBio Revio
Up to 360 Gb of HiFi reads per day, equivalent to 1,300 human whole genomes per year.
Tens of kilobases long HiFi reads. Price example (Sequel II): ~35kSEK per library and SMRT cell
Sequencing approaches
Despite price drop, still need to make choices regarding depth and breadth of sequencing coverage and number of samples.
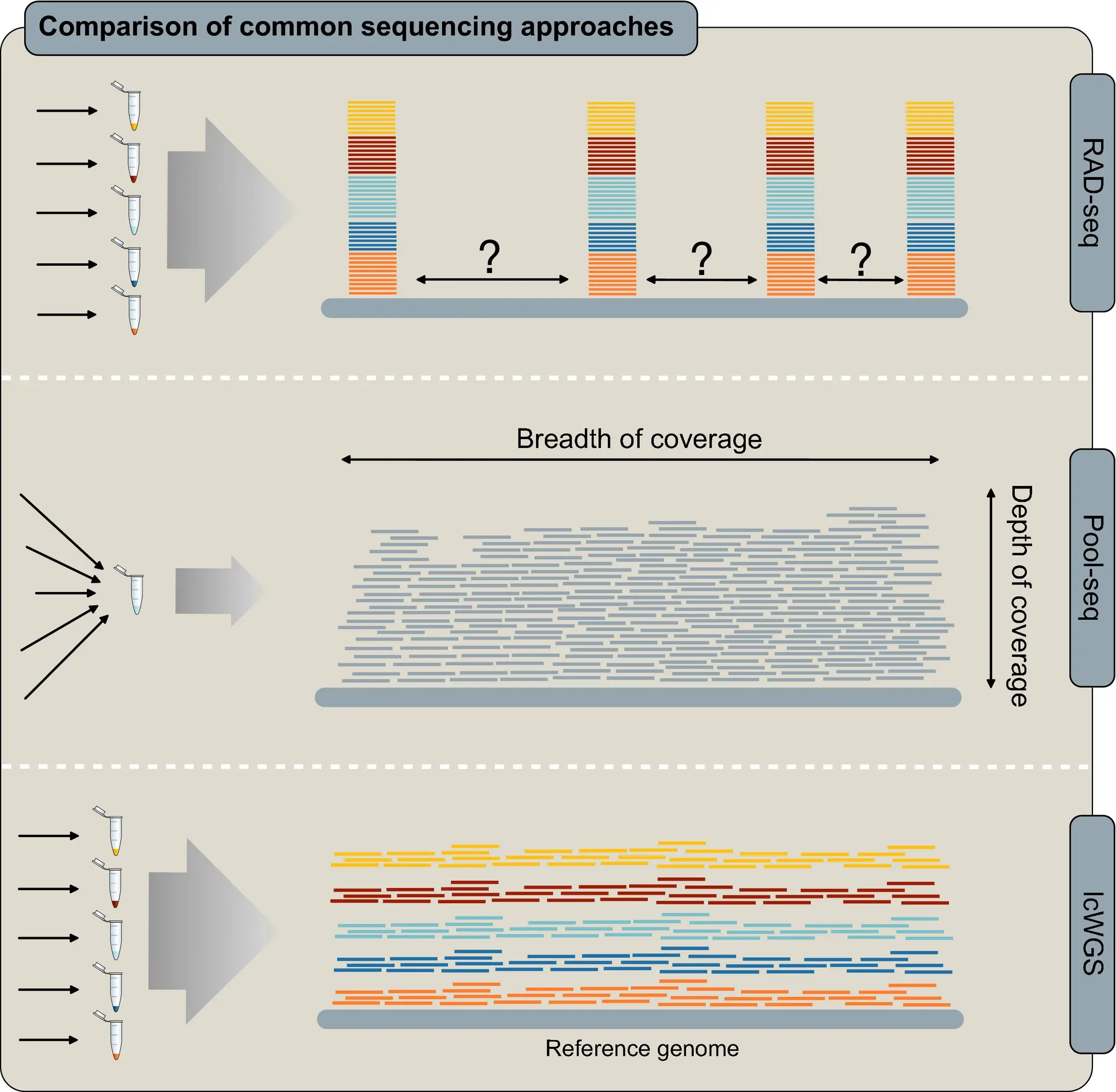
Lou et al. (2021)
Our focus will be on Whole Genome reSequencing (WGS), mostly high-coverage.
Genome assembly and population resequencing
Genome assembly
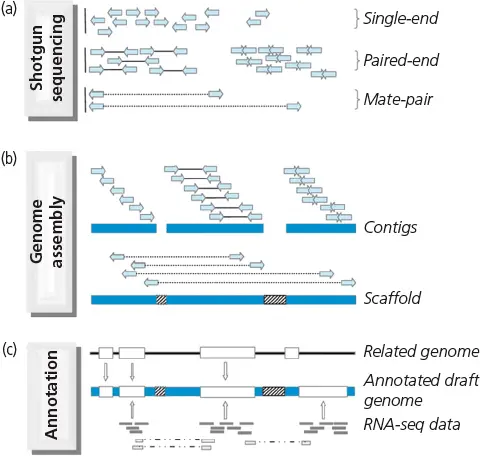
Allendorf et al. (2022)
Population resequencing
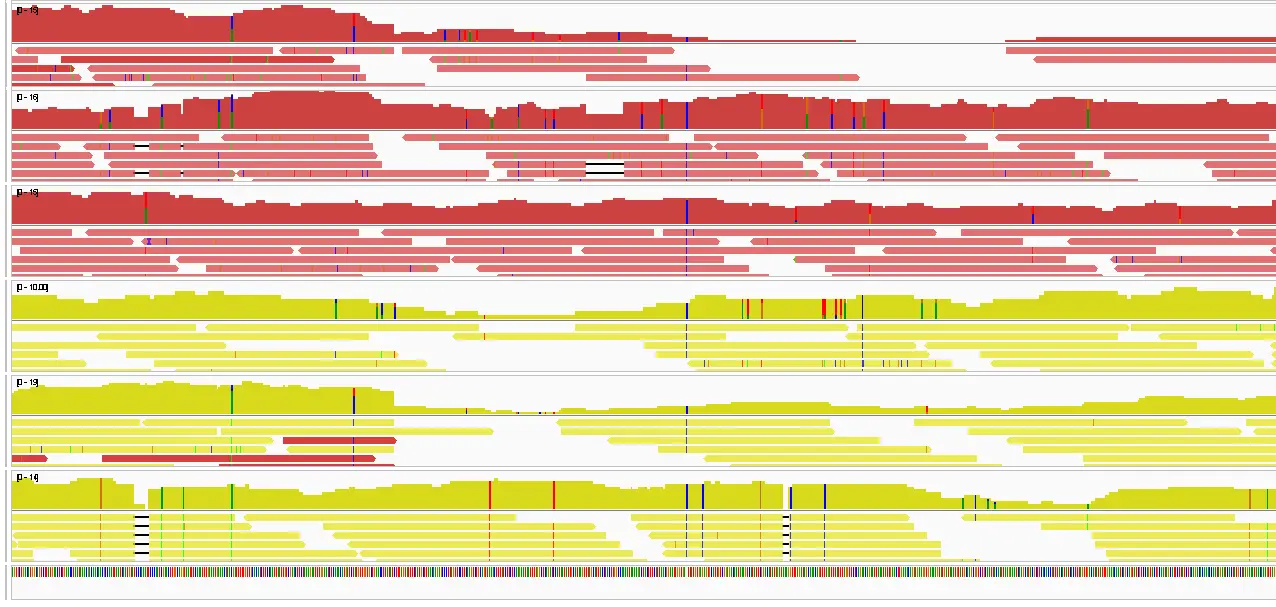
DNA sequence quality control
Quality values represent the probability P that the call is incorrect. They are coded as Phred quality scores Q. Here, Q=20 implies 1% probability of error, Q=30 implies 0.1% and so on. Typically you should not rely on quality values below 20.
Q = -10 \log_{10} P
A common way to do QC is with fastqc:
- For Illumina paired-end sequencing, the second read pair usually shows a larger drop in quality towards ends
- Trimming the sequences for adapter sequence and quality is good practice (e.g., with CutAdapt (Martin, 2011))

Bibliography
Allendorf, F. W., Funk, W. C., Aitken, S. N., Byrne, M., & Luikart, G. (2022). Population Genomics. In F. W. Allendorf, W. C. Funk, S. N. Aitken, M. Byrne, G. Luikart, & A. Antunes (Eds.), Conservation and the Genomics of Populations (p. 0). Oxford University Press. https://doi.org/10.1093/oso/9780198856566.003.0004
Lou, R. N., Jacobs, A., Wilder, A. P., & Therkildsen, N. O. (2021). A beginner’s guide to low-coverage whole genome sequencing for population genomics. Molecular Ecology, 30(23), 5966–5993. https://doi.org/10.1111/mec.16077
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, 17(1), pp. 10–12. https://doi.org/10.14806/ej.17.1.200
Wetterstrand, KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). www.genome.gov/sequencingcostsdata