Genetic diversity
What determines diversity levels?

The usual questions:
What evolutionary forces maintain genetic diversity in natural populations? How do diversity levels relate to census population sizes…? Do low levels of diversity limit adaptation to selective pressures?
Leffler et al. (2012)
After allozyme era, the study of genetic diversity was largely neglected due to lack of genome-wide data, but with advent of population genomics becoming a hot topic again.
Ellegren & Galtier (2016)
Lewontin’s paradox: genetic diversity range smaller than variation among species in population size
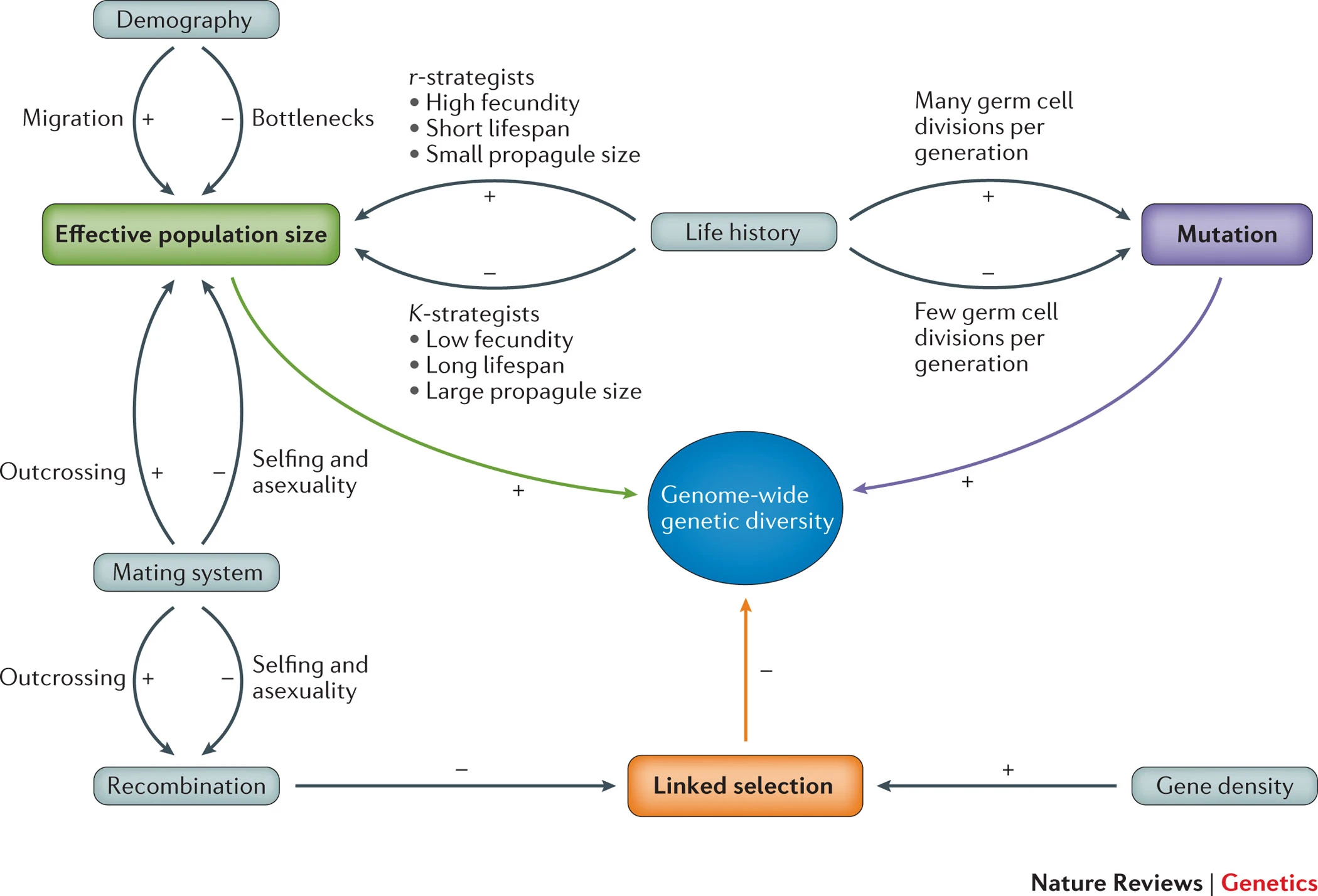
Factors that influence genetic diversity
Genetic drift
Reduces diversity at loss \propto \frac{1}{N}
Selection
Adaptive selection decreases variation, more so if acting on new mutations compared to standing variation.
Balancing selection may increase variation.
Recombination
Low recombination rates lead to less “reshuffling” of variation and hence lower diversity.
Genetic diversity in conservation biology
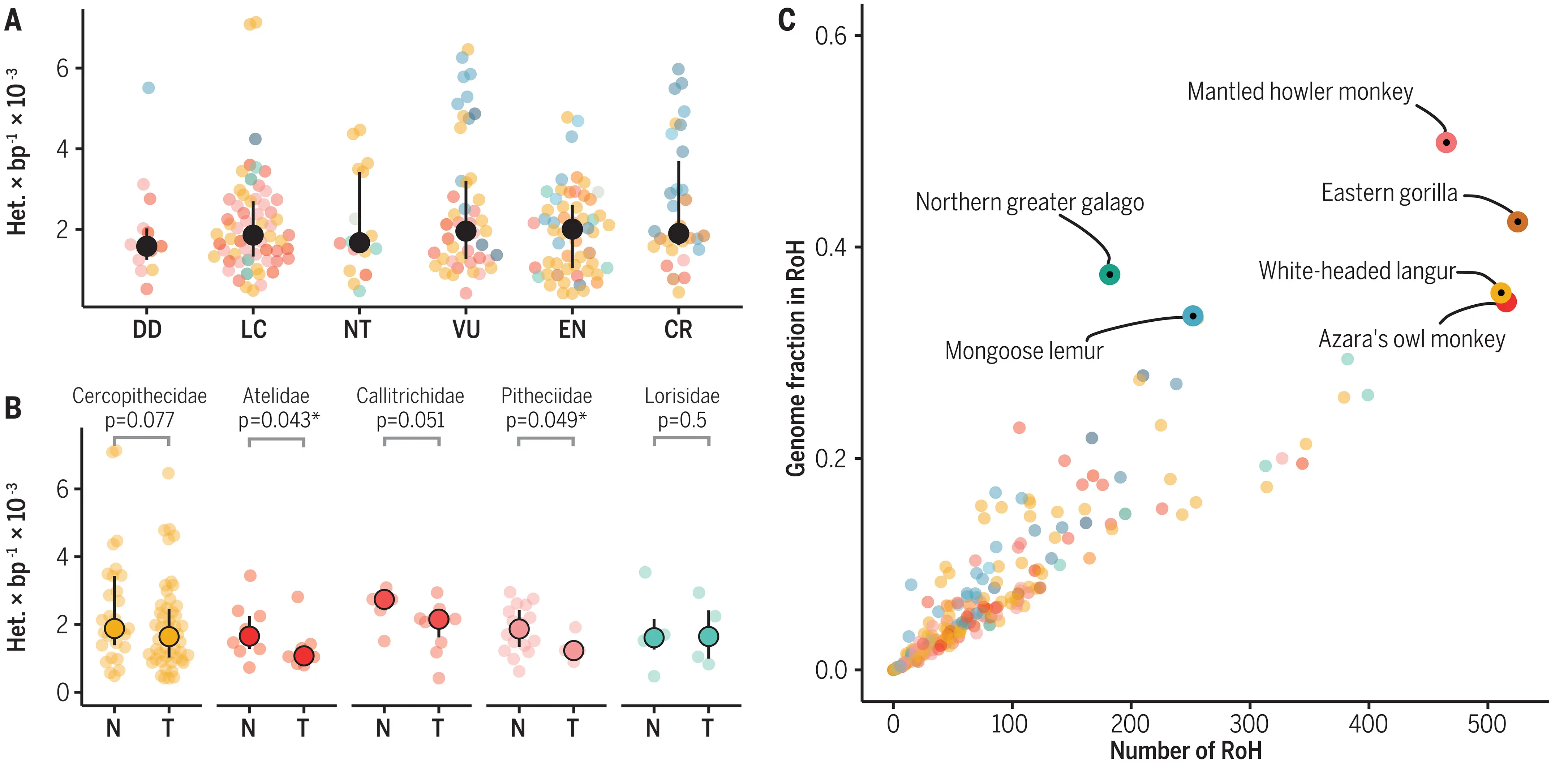
…no global relationship between numerically coded IUCN extinction risk categories and estimated heterozygosity…
Low genetic diversity symptom of past genetic drift inbreeding (higher levels of homozygosity), caused by low N_e
García-Dorado & Caballero (2021)
However: if population decline is rapid, may be too little time for inbreeding to occur \Rightarrow genetic diversity within species not necessarily aligned to extinction risk
Lewis (2023)
Many programs treat missing data as invariant

Diversity:
\pi = \frac{\sum_{i<j}k_{ij}}{n \choose 2}
Divergence:
d_{XY} = \frac{1}{n_Xn_Y}\sum_{i=1}^{n_X}\sum_{j=1}^{n_Y}k_{ij}
Here, n is the number of samples, k_{ij} tally of allelic differences between two haplotypes within (\pi) a population or between (d_{XY}) populations
Missing data may bias diversity measures downwards

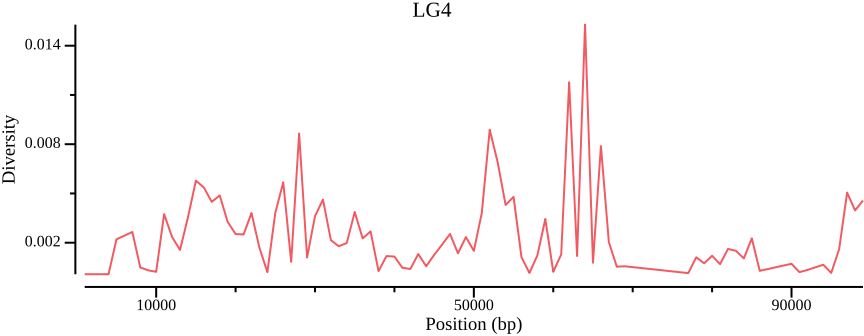
Nucleotide diversity landscapes
Genetic basis of adaptation and genome scans
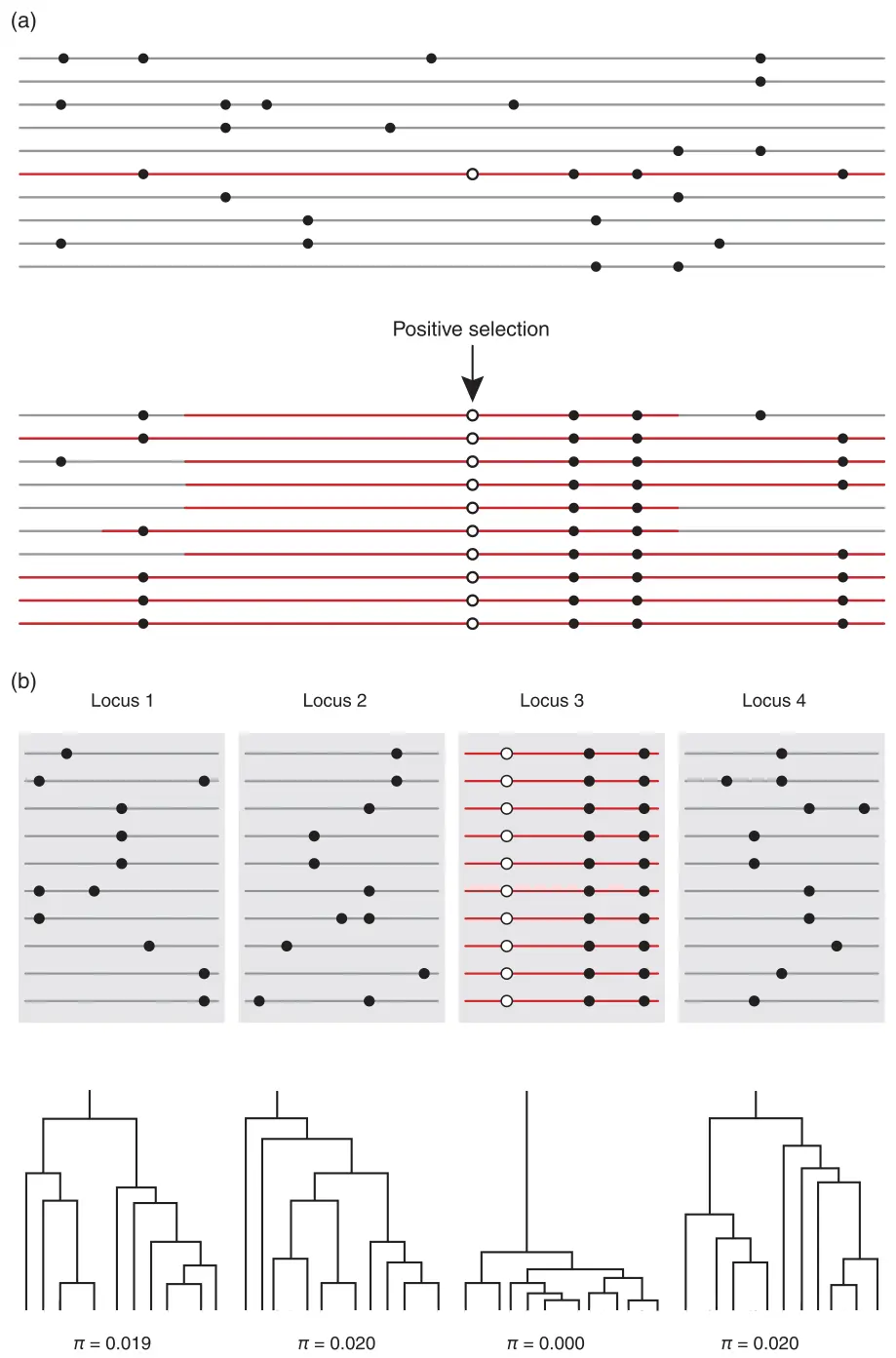
Fundamental questions:
- How many genes are involved in the evolution of adaptive traits?
- What is the distribution of phenotypic effects among successive allelic substitutions?
- Is adaptation typically based on standing variation or new mutations?
- What is the relative importance of additive vs. nonadditive effects on adaptive trait variation?
- And what is the relative importance of structural vs. regulatory changes in phenotypic evolution?
Storz (2005), Fig 1
Example
vcftools --gzvcf vcf/allsites.vcf.gz --weir-fst-pop PUN-Y.txt \
--weir-fst-pop PUN-R.txt \
--fst-window-size 1000 \
--out results/out
csvtk plot line --tabs results/out.windowed.weir.fst \
-x BIN_START -y MEAN_FST \
--point-size 2 --xlab "Position (bp)" \
--ylab "Fst" --title "LG4: PUN-Y vs PUN-R" \
--width 9.0 --height 3.5 --scatter \
> results/out.windowed.weir.fst.mean.png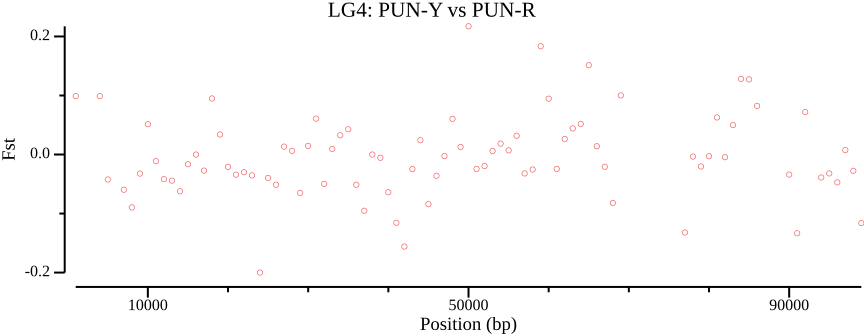
Z-scores can help identifying outliers

Raw data can be converted to Z-scores to highlight outliers. A Z-score is a measure of how far a data point is from the mean in terms of the number of standard deviations:
Z = \frac{X - \mu}{\sigma}
Threshold of a couple of standard deviations common.
LD decay and choice of window size

Properties of genetic variation and inferred demographic history in sampled A. millepora. Fuller et al. (2020), Figure 2. Upper left plot illustrates LD as a function of physical distance. Here, choosing a window size 20-30kb would ensure that most windows are independent.
Dissecting differentiation landscapes
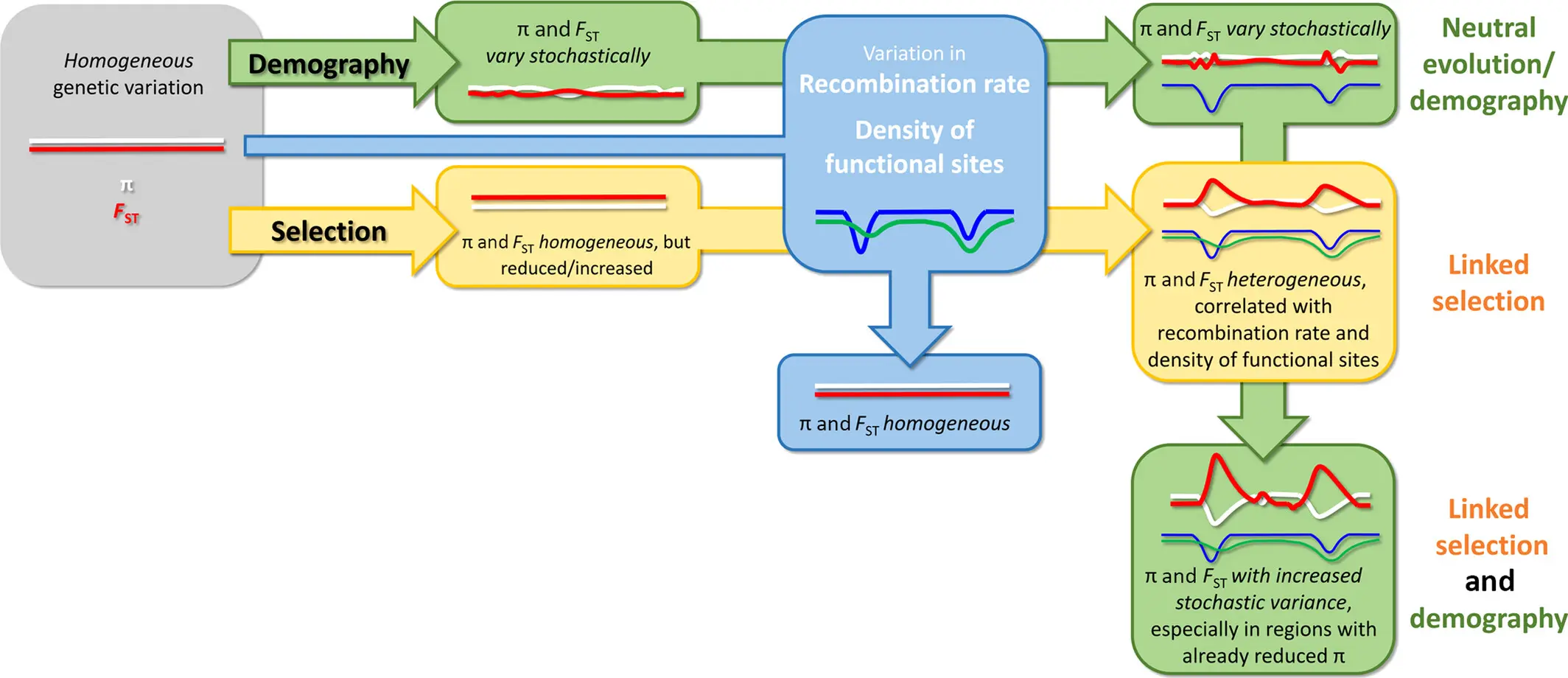
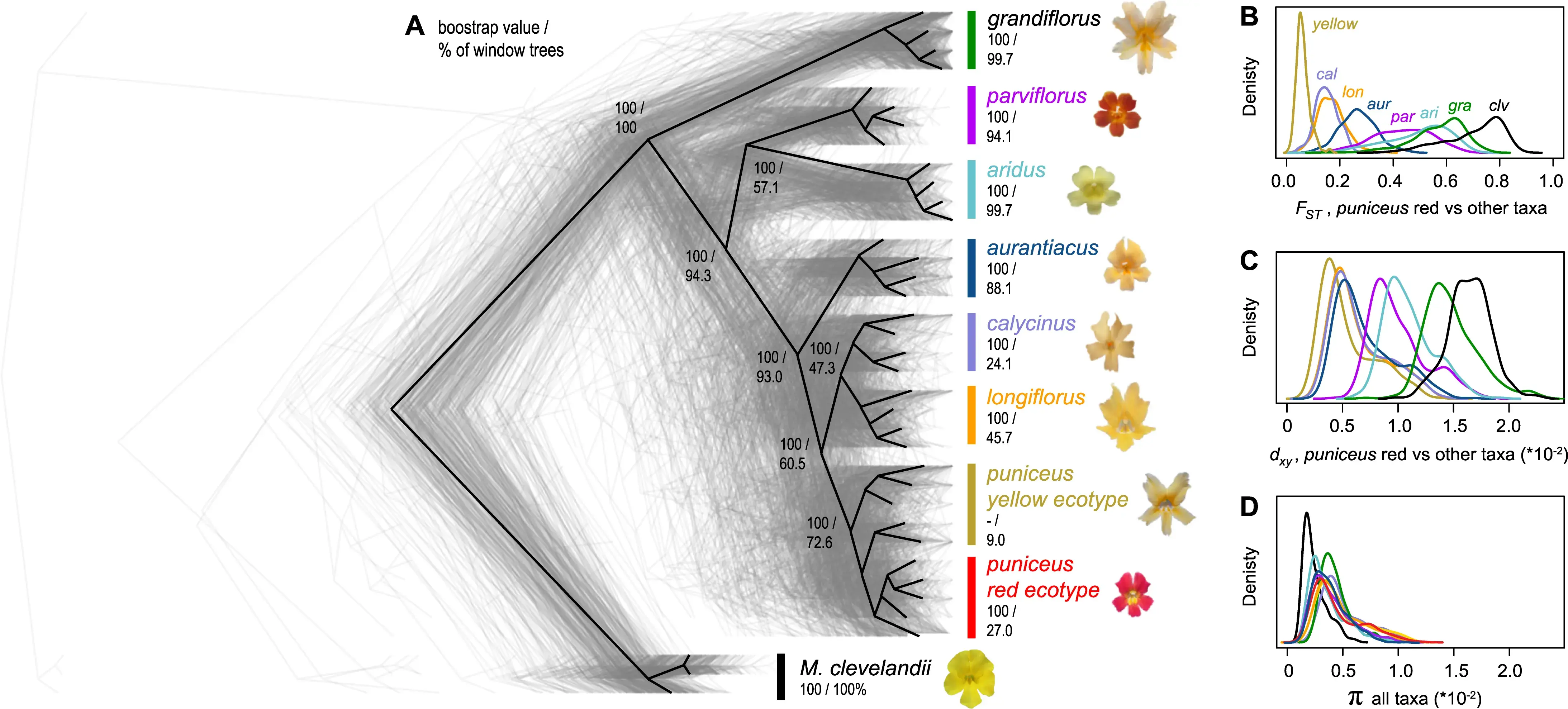
Monkeyflower genomic landscape
Bibliography
Berner, D. (2019). Allele Frequency Difference AFD to FST for Quantifying Genetic Population Differentiation. Genes, 10(4), 308. https://doi.org/10.3390/genes10040308
Burri, R. (2017). Dissecting differentiation landscapes: A linked selection’s perspective. Journal of Evolutionary Biology, 30(8), 1501–1505. https://doi.org/10.1111/jeb.13108
Charlesworth, B., & Jensen, J. D. (2022). How Can We Resolve Lewontin’s Paradox? Genome Biology and Evolution, 14(7), evac096. https://doi.org/10.1093/gbe/evac096
Corbett-Detig, R. B., Hartl, D. L., & Sackton, T. B. (2015). Natural Selection Constrains Neutral Diversity across A Wide Range of Species. PLOS Biology, 13(4), e1002112. https://doi.org/10.1371/journal.pbio.1002112
Ellegren, H., & Galtier, N. (2016). Determinants of genetic diversity. Nature Reviews Genetics, 17(7), 422–433. https://doi.org/10.1038/nrg.2016.58
Fuller, Z. L., Mocellin, V. J. L., Morris, L. A., Cantin, N., Shepherd, J., Sarre, L., Peng, J., Liao, Y., Pickrell, J., Andolfatto, P., Matz, M., Bay, L. K., & Przeworski, M. (2020). Population genetics of the coral Acropora millepora: Toward genomic prediction of bleaching. Science, 369(6501), eaba4674. https://doi.org/10.1126/science.aba4674
García-Dorado, A., & Caballero, A. (2021). Neutral genetic diversity as a useful tool for conservation biology. Conservation Genetics, 22(4), 541–545. https://doi.org/10.1007/s10592-021-01384-9
Korunes, K. L., & Samuk, K. (2021). Pixy: Unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Molecular Ecology Resources, 21(4), 1359–1368. https://doi.org/10.1111/1755-0998.13326
Kuderna, L. F. K., Gao, H., Janiak, M. C., Kuhlwilm, M., Orkin, J. D., Bataillon, T., Manu, S., Valenzuela, A., Bergman, J., Rousselle, M., Silva, F. E., Agueda, L., Blanc, J., Gut, M., de Vries, D., Goodhead, I., Harris, R. A., Raveendran, M., Jensen, A., … Marques Bonet, T. (2023). A global catalog of whole-genome diversity from 233 primate species. Science, 380(6648), 906–913. https://doi.org/10.1126/science.abn7829
Leffler, E. M., Bullaughey, K., Matute, D. R., Meyer, W. K., Ségurel, L., Venkat, A., Andolfatto, P., & Przeworski, M. (2012). Revisiting an Old Riddle: What Determines Genetic Diversity Levels within Species? PLOS Biology, 10(9), e1001388. https://doi.org/10.1371/journal.pbio.1001388
Lewis, D. (2023). Biggest ever study of primate genomes has surprises for humanity. Nature. https://doi.org/10.1038/d41586-023-01776-6
Nei, M. (1973). Analysis of Gene Diversity in Subdivided Populations. Proceedings of the National Academy of Sciences, 70(12), 3321–3323. https://doi.org/10.1073/pnas.70.12.3321
Stankowski, S., Chase, M. A., Fuiten, A. M., Rodrigues, M. F., Ralph, P. L., & Streisfeld, M. A. (2019). Widespread selection and gene flow shape the genomic landscape during a radiation of monkeyflowers. PLOS Biology, 17(7), e3000391. https://doi.org/10.1371/journal.pbio.3000391
Storz, J. F. (2005). INVITED REVIEW: Using genome scans of DNA polymorphism to infer adaptive population divergence. Molecular Ecology, 14(3), 671–688. https://doi.org/10.1111/j.1365-294X.2005.02437.x
Wright, S. (1931). Evolution in Mendelian Populations. Genetics, 16(2), 97–159. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1201091/