
Genetic diversity
The origin and maintenance of genetic diversity
Origin and change of variation
Mutation
Selection
Recombination
Drift
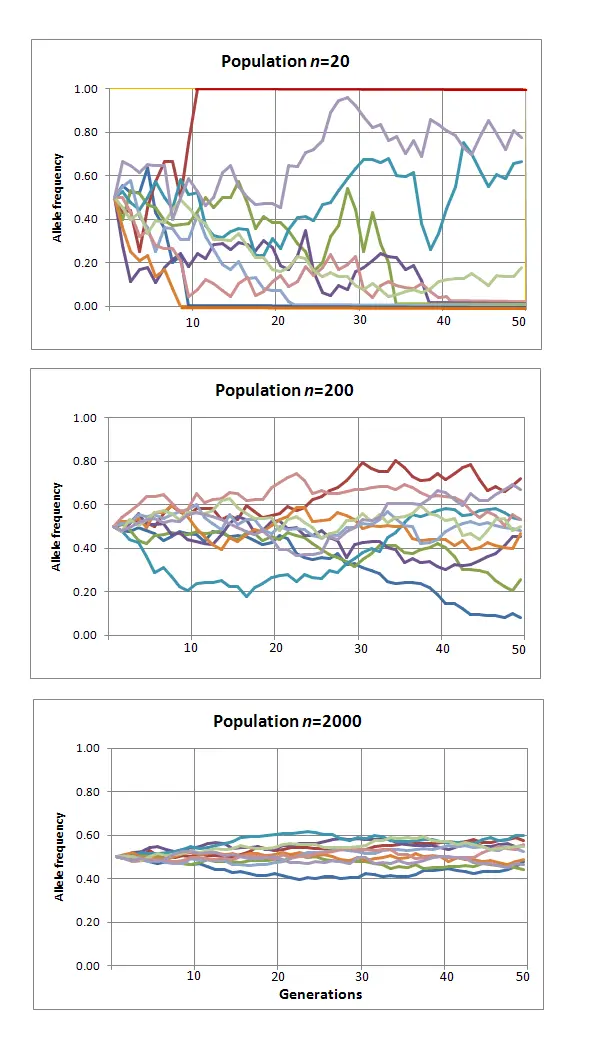
Wright-Fisher model with alleles

Alleles can randomly fix or be lost through process called genetic drift
Wright-Fisher model showing the evolution of population of 10 genes over 16 generations. Allele variants are shown in white and black. Starting frequency black variant is 0.3.
Binomial process models allele sampling

We assume two alleles A, a, each with i and j=2N-i copies in generation t.
![]()
![]()
![]()
![]()
![]()
![]()
i=8, j=2\cdot 6-8=4
Let p_t=i/2N be the frequency of A in generation t, and q_t=1-p_t the frequency of a.
![]()
![]()
![]()
![]()
![]()
![]()
p_t = 8/12
![]()
![]()
![]()
![]()
![]()
![]()
p_{t+1} = 4/12
Prob(k A alleles in next generation) is \mathsf{Bin}(2N, \frac{i}{2N})
Genetic drift
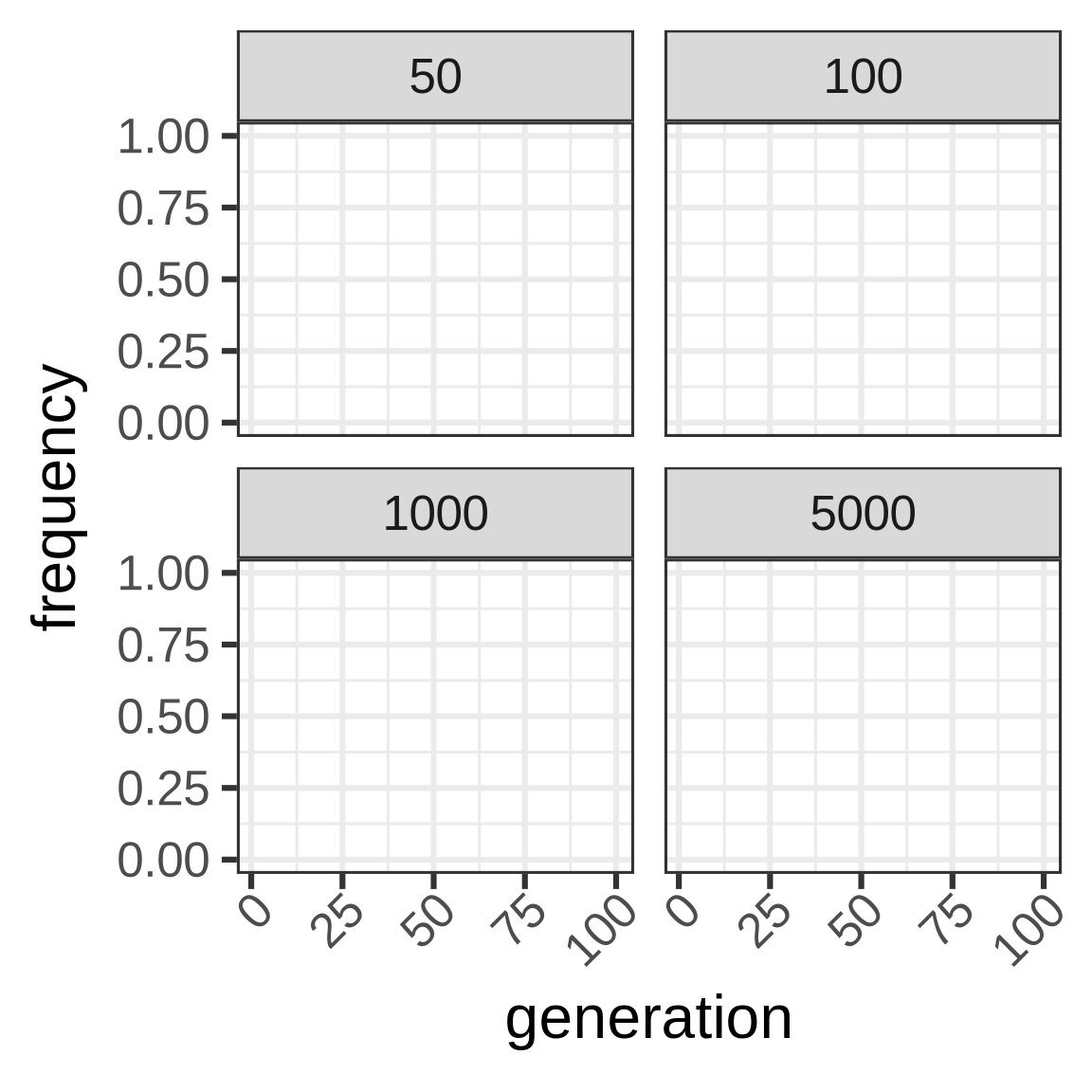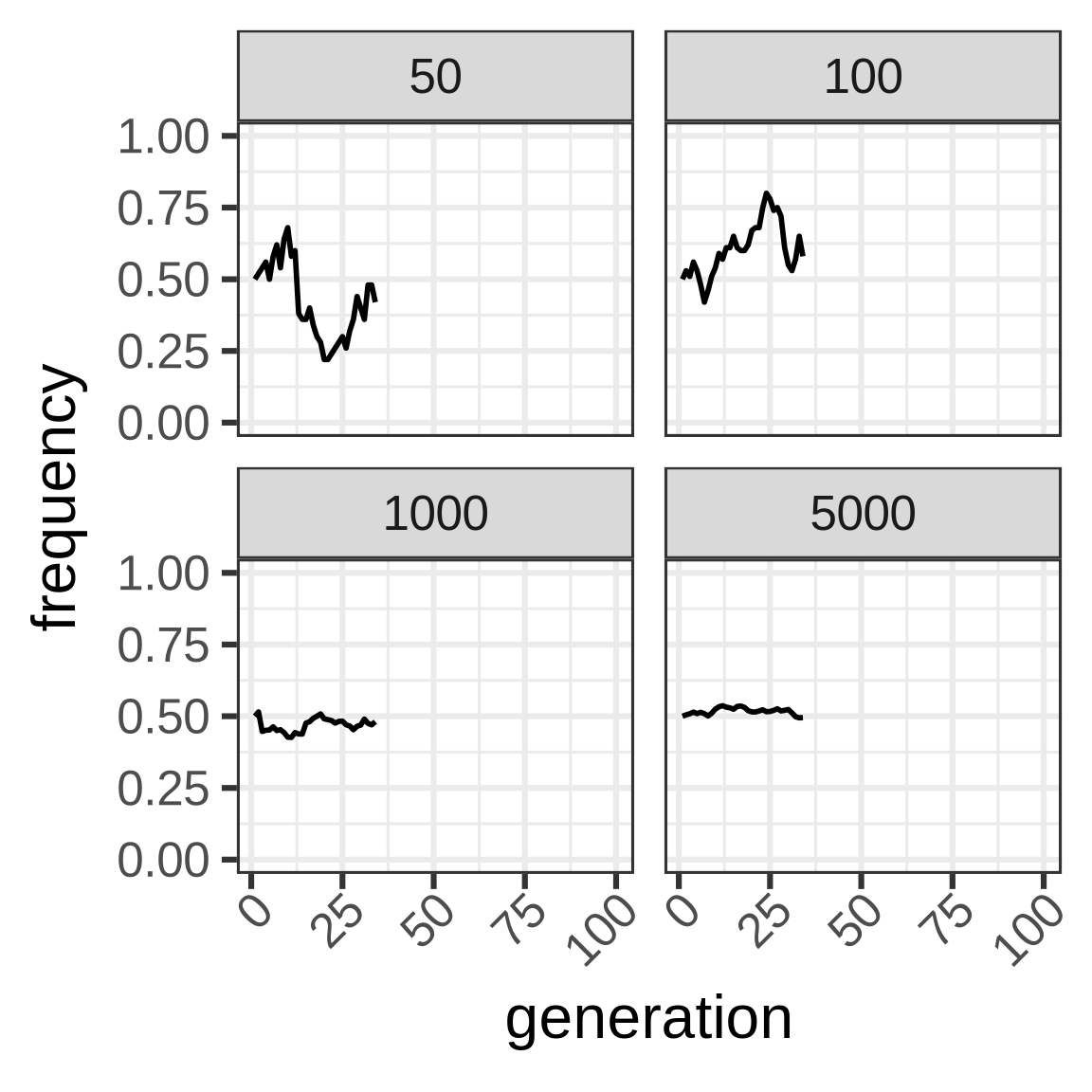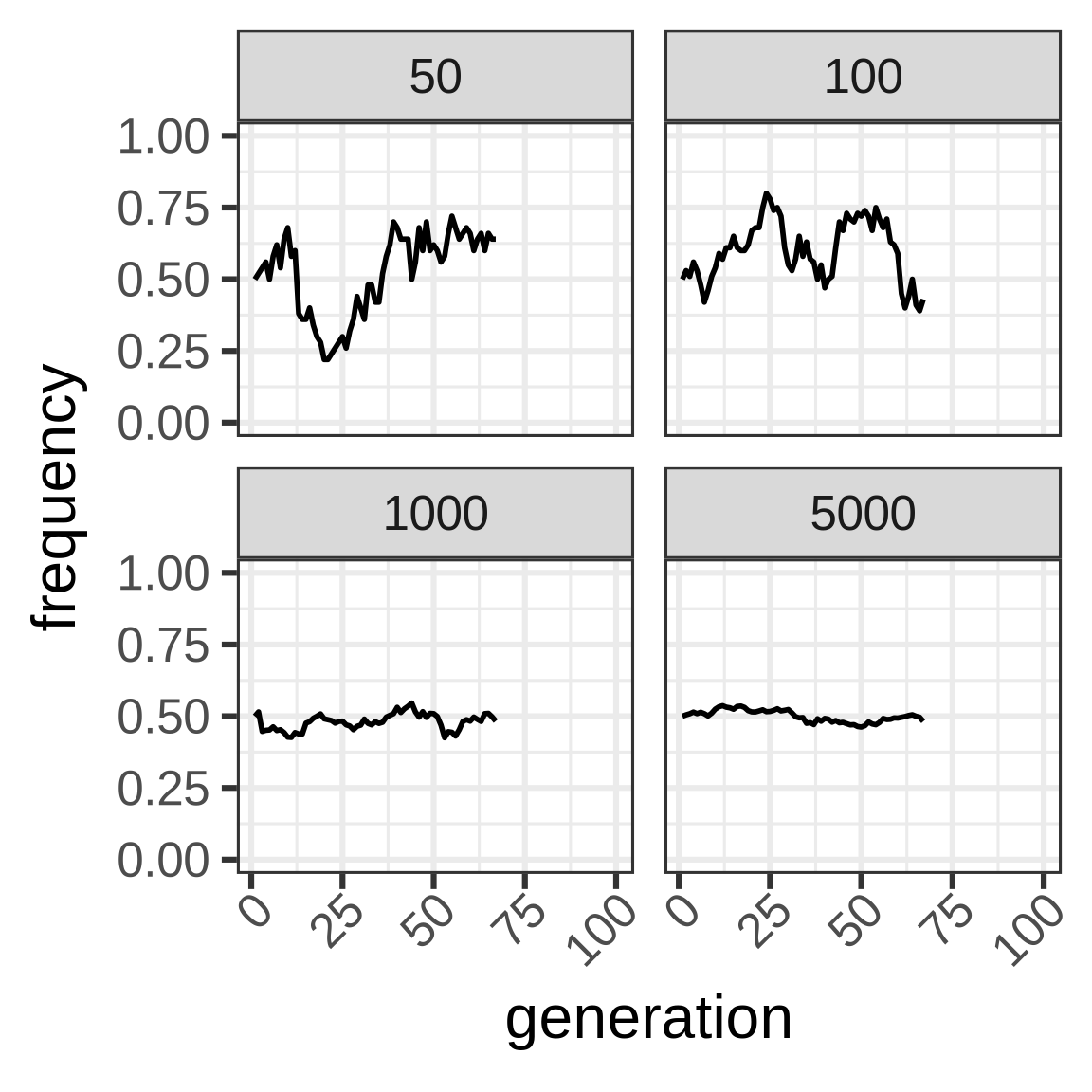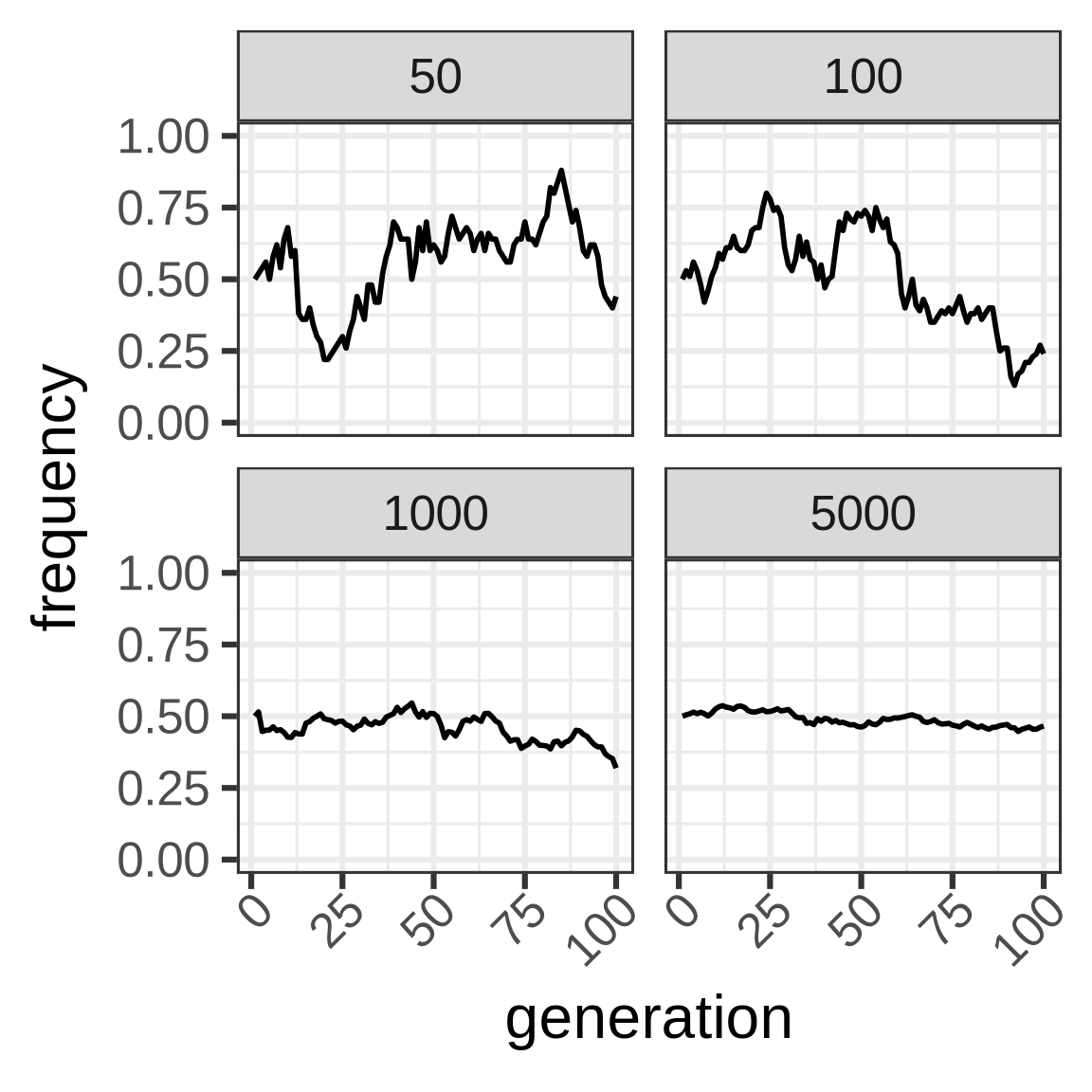
To capture dynamics, follow allele frequency trajectory (p_t) as function of time.
##' Wright Fisher model - follow allele frequency distribution
##'
##' @param p0 Starting frequency
##' @param n Population size
##' @param generations Number of generations to simulate
##'
wright_fisher <- function(p0, n, generations) {
x <- vector(mode = "numeric", length = generations)
x[1] <- p0
for (i in seq(2, length(x))) {
x[i] <- rbinom(1, size = n, prob = x[i - 1])/n
}
x
}
Genetic drift

- fate of allele: fixation or loss \rightarrow eventually loss of variation
- probability of fixation \pi(p)=p, where p is the current frequency
- rate of drift (loss of variation) \propto \frac{1}{2N}

Allele frequency distribution for N=1
Instead of looking at frequencies let’s switch to distributions of alleles for one individual, one locus. Then there are three possible genotypes (states) aa, aA, and AA. Let n=0,1,2 be an integer corresponding to each genotype (i.e., it counts the number of A alleles).
Assume individual mates with itself at random(!) starting in either of the three states. How does distribution evolve?
t=0



t=1



t=2

Probability distributions of allele frequencies


Mathematical treatment of drift can become complicated: easier to study dynamics of heterozygosity
Heterozygosity dynamics

Let \mathcal{H}_t be the probability that two alleles are different by state. One can show that the time course evolution of \mathcal{H}_t in a randomly mating population consisting of N diploid hermaphroditic individuals is
\mathcal{H}_t = \mathcal{H}_0 \left( 1 - \frac{1}{2N} \right)^t
Important consequence: heterozygosity in WF population lost at rate 1/2N.
Heterozygosity dynamics



Example of how rapid decline in population size can affect heterozygosity.
Population size influences genetic diversity!
However, census population size not (always) the correct measure.
Mutation
|
|
|


- Two-allele
- Derive popgen stats
- Finite sites
- Recurrent mutations
- Infinite alleles
- Protein electrophoresis
- Inifinite sites
- DNA sequences
Mutation and drift
Genetic drift “moves” frequencies to the point that variation is lost via allele fixation or loss. New variation is introduced through mutation. We typically assume mutations are described by a Poisson process with rate \mu (per generation).
The mutation rate is denoted \mu, and the population scaled mutation rate is 2N_e\mu for haploid populations, 4N_e\mu for diploid, where N_e is the effective population size.
The mutation - drift balance is when the diversity lost due to drift equals the diversity gained due to mutation.

Tracing the evolution of mutations

Observation: most mutations are in fact lost
Recall: fixation probability \pi(p)=p
The neutral theory of evolution
Mutation drift balance, together with the observation during 50’s-60’s that polymorphism was more common than expected, is the foundation of the neutral theory of evolution (Kimura, 1983): allele frequencies may change and fix due to chance alone and not selection; most mutations behave as if they are neutral.
Nearly neutral theory (Ohta, 1973) was later developed to explain failure to predict scaling of polymorphism with population size: most mutations are not neutral but slightly deleterious and purged from population by natural selection.

Bibliography
Barton, N. H., Briggs, D. E. G., Eisen, J. A., Goldstein, D. B., & Patel, N. H. (2007). Evolution. Cold Spring Harbor Laboratory Press.
Buri, P. (1956). Gene Frequency in Small Populations of Mutant Drosophila. Evolution, 10(4), 367–402. https://doi.org/10.1111/j.1558-5646.1956.tb02864.x
Charlesworth, B., & Charlesworth, D. (2010). Elements of Evolutionary Genetics. Roberts and Company Publishers.
Ewens, W. J. (2004). Mathematical Population Genetics (S. S. Antman, J. E. Marsden, L. Sirovich, & S. Wiggins, Eds.; Vol. 27). Springer. https://doi.org/10.1007/978-0-387-21822-9
Gillespie, J. H. (2004). Population Genetics: A Concise Guide (2nd edition). Johns Hopkins University Press.
Graham Coop. (2020). Notes on Population Genetics. https://github.com/cooplab/popgen-notes
Hubisz, M., & Siepel, A. (2020). Inference of Ancestral Recombination Graphs Using ARGweaver. In J. Y. Dutheil (Ed.), Statistical Population Genomics (pp. 231–266). Springer US. https://doi.org/10.1007/978-1-0716-0199-0_10
Hurst, L. D. (2009). Genetics and the understanding of selection. Nature Reviews Genetics, 10(2), 83–93. https://doi.org/10.1038/nrg2506
Kimura, M. (1983). The neutral theory of molecular evolution. Cambridge University Press. https://doi.org/10.1017/CBO9780511623486
Kimura, M., & Ohta, T. (1971). Protein Polymorphism as a Phase of Molecular Evolution. Nature, 229(5285), 467–469. https://doi.org/10.1038/229467a0
Kumar, S., & Subramanian, S. (2002). Mutation rates in mammalian genomes. Proceedings of the National Academy of Sciences, 99(2), 803–808. https://doi.org/10.1073/pnas.022629899
Leffler, E. M., Bullaughey, K., Matute, D. R., Meyer, W. K., Ségurel, L., Venkat, A., Andolfatto, P., & Przeworski, M. (2012). Revisiting an Old Riddle: What Determines Genetic Diversity Levels within Species? PLOS Biology, 10(9), e1001388. https://doi.org/10.1371/journal.pbio.1001388
Ohta, T. (1973). Slightly Deleterious Mutant Substitutions in Evolution. Nature, 246(5428), 96. https://doi.org/10.1038/246096a0
Waples, R. S. (2022). What Is Ne, Anyway? Journal of Heredity, 113(4), 371–379. https://doi.org/10.1093/jhered/esac023
Waples, R. S. (2025). The Idiot’s Guide to Effective Population Size. Molecular Ecology, e17670. https://doi.org/10.1111/mec.17670
Wisely, S. M., Buskirk, S. W., Fleming, M. A., McDonald, D. B., & Ostrander, E. A. (2002). Genetic Diversity and Fitness in Black-Footed Ferrets Before and During a Bottleneck. Journal of Heredity, 93(4), 231–237. https://doi.org/10.1093/jhered/93.4.231