class: center, middle, inverse, title-slide # Analysis of bulk RNA-Seq data ## Introduction To Bioinformatics Using NGS Data ### 25-May-2021 ### NBIS --- exclude: true count: false <link href="https://fonts.googleapis.com/css?family=Roboto|Source+Sans+Pro:300,400,600|Ubuntu+Mono&subset=latin-ext" rel="stylesheet"> <link rel="stylesheet" href="https://use.fontawesome.com/releases/v5.3.1/css/all.css" integrity="sha384-mzrmE5qonljUremFsqc01SB46JvROS7bZs3IO2EmfFsd15uHvIt+Y8vEf7N7fWAU" crossorigin="anonymous"> <!-- ------------ Only edit title, subtitle & author above this ------------ --> --- name: contents class: spaced ## Contents * [RNA Sequencing](#intro) * [Workflow](#workflow) * [DGE Workflow](#workflow-dge) * [ReadQC](#read-qc) * [Mapping](#mapping-intro) * [Alignment QC](#alignment-qc) * [Quantification](#quantification-counts) * [Normalisation](#normalisation) * [Exploratory](#exploratory-heatmap) * [DGE](#dge-1) * [Functional analyses](#functional-analysis-1) * [Summary](#summary) * [Help](#help) --- name: intro class: spaced ## RNA Sequencing  - The transcriptome is spatially and temporally dynamic - Data comes from functional units (coding regions) - Only a tiny fraction of the genome ??? * What is RNA? * Central dogma of molecular biology. DNA -> RNA -> Protein. * We have gene models to explain organisation of the DNA into functional units. * Very tiny portion of the genome transcribes to RNA. * There are many types of RNA. Commonly protein coding RNA or mRNA. There are tRNA, sRNA, miRNA, siRNA, lincRNA etc. * What is the most abundant RNA? rRNA. * Why are we interested in RNA? While DNA is constant in all cells, expressed RNA varies from cell to cell and from time to time. * When we say RNA sequencing, what we sequencing?. For most part we are converting RNA to DNA and sequencing the DNA just like regular DNA sequencing. --- name: applications class: spaced ## Applications - Identify gene sequences in genomes - Learn about gene function - Differential gene expression - Explore isoform and allelic expression - Understand co-expression, pathways and networks - Gene fusion - RNA editing --- name: workflow class: spaced ## Workflow .size-80[] <img src="data/rnaseq/sequence.jpg" style="height:250px; position:fixed; right:0px; bottom:0px; margin-right: 100px; margin-bottom: 130px; border-radius:4px;"> .citation[ .cite[<i class="fas fa-link"></i> Conesa, Ana, *et al.* "A survey of best practices for RNA-seq data analysis." [Genome biology 17.1 (2016): 13](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8)] ] --- name: exp-design class: spaced ## Experimental design .pull-left-50[ - Balanced design - Technical replicates not necessary (.altcol[Marioni *et al.*, 2008]) - Biological replicates: 6 - 12 (.altcol[Schurch *et al.*, 2016]) - ENCODE consortium - Previous publications - Power analysis <i class="fas fa-toolbox"></i> [RnaSeqSampleSize](https://cqs.mc.vanderbilt.edu/shiny/RnaSeqSampleSize/) (Power analysis), [Scotty](http://scotty.genetics.utah.edu/) (Power analysis with cost) ] .pull-right-50[ .size-90[] ] .citation[ .cite[<i class="fas fa-link"></i> Busby, Michele A., *et al.* "Scotty: a web tool for designing RNA-Seq experiments to measure differential gene expression." [Bioinformatics 29.5 (2013): 656-657](https://academic.oup.com/bioinformatics/article/29/5/656/252753)] .cite[<i class="fas fa-link"></i> Marioni, John C., *et al.* "RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays." [Genome research (2008)](https://genome.cshlp.org/content/18/9/1509.long)] .cite[<i class="fas fa-link"></i> Hart, S. N., Therneau, T. M., Zhang, Y., Poland, G. A., & Kocher, J. P. (2013). Calculating sample size estimates for RNA sequencing data. [Journal of computational biology, 20(12), 970-978](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3842884/).] .cite[<i class="fas fa-link"></i> Schurch, Nicholas J., *et al.* "How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use?." [Rna (2016)](http://rnajournal.cshlp.org/content/early/2016/03/30/rna.053959.115.abstract)] .cite[<i class="fas fa-link"></i> Zhao, Shilin, *et al.* "RnaSeqSampleSize: real data based sample size estimation for RNA sequencing." [BMC bioinformatics 19.1 (2018): 191](https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2191-5)] ] --- name: rna-extraction class: spaced ## RNA extraction .pull-left-50[ - Sample processing and storage - Total RNA/mRNA/small RNA - DNAse treatment - Quantity & quality - RIN values (Strong effect) - Batch effect - Extraction method bias (GC bias) ] .pull-right-50[ .size-70[] ] .citation[ .cite[<i class="fas fa-link"></i> Romero, Irene Gallego, *et al*. "RNA-seq: impact of RNA degradation on transcript quantification." [BMC biology 12.1 (2014): 42](https://bmcbiol.biomedcentral.com/articles/10.1186/1741-7007-12-42)] .cite[<i class="fas fa-link"></i> Kim, Young-Kook, *et al*. "Short structured RNAs with low GC content are selectively lost during extraction from a small number of cells." [Molecular cell 46.6 (2012): 893-895](https://www.cell.com/molecular-cell/fulltext/S1097-2765(12)00481-9).] ] --- name: library-prep class: spaced ## Library prep .pull-left-50[ - PolyA selection - rRNA depletion - Size selection - PCR amplification (See section PCR duplicates) - Stranded (directional) libraries - Accurately identify sense/antisense transcript - Resolve overlapping genes - Exome capture - Library normalisation - Batch effect ] .pull-right-50[  ] .citation[ .cite[<i class="fas fa-link"></i> Zhao, Shanrong, et al. "Comparison of stranded and non-stranded RNA-seq transcriptome profiling and investigation of gene overlap." [BMC genomics 16.1 (2015): 675](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4559181/)] .cite[<i class="fas fa-link"></i> Levin, Joshua Z., et al. "Comprehensive comparative analysis of strand-specific RNA sequencing methods." [Nature methods 7.9 (2010): 709](https://www.nature.com/articles/nmeth.1491)] ] --- name: sequencing ## Sequencing .pull-left-60[ - Sequencer (Illumina/PacBio) - Read length - Greater than 50bp does not improve DGE - Longer reads better for isoforms - Pooling samples - Sequencing depth (Coverage/Reads per sample) - Single-end reads (Cheaper) - Paired-end reads - Increased mappable reads - Increased power in assemblies - Better for structural variation and isoforms - Decreased false-positives for DGE ] .pull-right-40[  ] .citation[ .cite[<i class="fas fa-link"></i> Chhangawala, Sagar, et al. "The impact of read length on quantification of differentially expressed genes and splice junction detection." [Genome biology 16.1 (2015): 131](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4531809/)] .cite[<i class="fas fa-link"></i> Corley, Susan M., et al. "Differentially expressed genes from RNA-Seq and functional enrichment results are affected by the choice of single-end versus paired-end reads and stranded versus non-stranded protocols." [BMC genomics 18.1 (2017): 399](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5442695/)] .cite[<i class="fas fa-link"></i> Liu, Yuwen, Jie Zhou, and Kevin P. White. "RNA-seq differential expression studies: more sequence or more replication?." [Bioinformatics 30.3 (2013): 301-304](https://academic.oup.com/bioinformatics/article/30/3/301/228651)] .cite[<i class="fas fa-link"></i> Comparison of PE and SE for RNA-Seq, [SciLifeLab](https://ngisweden.scilifelab.se/file/1540-1_Comparison_of_PE_and_SE_for_RNA-seq.pdf)] ] --- name: workflow-dge ## Workflow • DGE  --- name: de-novo-assembly class: spaced ## De-Novo assembly - When no reference genome available - To identify novel genes/transcripts/isoforms - Identify fusion genes - Assemble transcriptome from short reads - Access quality of assembly and refine - Map reads back to assembled transcriptome <i class="fas fa-toolbox"></i> [Trinity](https://github.com/trinityrnaseq/trinityrnaseq/wiki), [SOAPdenovo-Trans](http://soap.genomics.org.cn/SOAPdenovo-Trans.html), [Oases](https://www.ebi.ac.uk/~zerbino/oases/), [rnaSPAdes](http://cab.spbu.ru/software/rnaspades/) .citation[ .cite[<i class="fas fa-link"></i> Hsieh, Ping-Han *et al*., "Effect of de novo transcriptome assembly on transcript quantification" [2018 bioRxiv 380998](https://www.biorxiv.org/content/early/2018/08/22/380998)] .cite[<i class="fas fa-link"></i> Wang, Sufang, and Michael Gribskov. "Comprehensive evaluation of de novo transcriptome assembly programs and their effects on differential gene expression analysis." [Bioinformatics 33.3 (2017): 327-333](https://academic.oup.com/bioinformatics/article/33/3/327/2580374)] ] --- name: read-qc ## Read QC - Number of reads - Per base sequence quality - Per sequence quality score - Per base sequence content - Per sequence GC content - Per base N content - Sequence length distribution - Sequence duplication levels - Overrepresented sequences - Adapter content - Kmer content <img src="data/rnaseq/qc.jpg" style="height:250px; position:fixed; right:0px; bottom: 0px; margin-right: 100px; margin-bottom: 310px;"> <i class="fas fa-toolbox"></i> [FastQC](https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), [MultiQC](http://multiqc.info/) https://sequencing.qcfail.com/  --- name: fastqc ## FastQC Good quality .size-50[[](http://www.bioinformatics.babraham.ac.uk/projects/fastqc/good_sequence_short_fastqc.html)] Poor quality .size-50[[](http://www.bioinformatics.babraham.ac.uk/projects/fastqc/bad_sequence_fastqc.html)] --- name: read-qc-2 ## Read QC • PBSQ, PSQS .size-85[.small[**Per base sequence quality**] ] .size-85[.small[**Per sequence quality scores**] ] --- name: read-qc-3 ## Read QC • PBSC, PSGC .size-85[.small[**Per base sequence content**] ] .size-85[.small[**Per sequence GC content**] ] --- name: read-qc-4 ## Read QC • SDL, AC .size-85[.small[**Sequence duplication level**] ] .size-85[.small[**Adapter content**] ] --- name: trimming ## Trimming .pull-left-50[ - Trim IF necessary - Synthetic bases can be an issue for SNP calling - Insert size distribution may be more important for assemblers - Trim/Clip/Filter reads - Remove adapter sequences - Trim reads by quality - Sliding window trimming - Filter by min/max read length - Remove reads less than ~18nt - Demultiplexing/Splitting <i class="fas fa-toolbox"></i> [Cutadapt](https://github.com/marcelm/cutadapt/), [fastp](https://github.com/OpenGene/fastp), [Skewer](https://github.com/relipmoc/skewer), [Prinseq](http://prinseq.sourceforge.net/) ] .pull-right-50[ .size-90[] ] --- name: mapping-intro ## Mapping  - Aligning reads back to a reference sequence - Mapping to genome vs transcriptome - Splice-aware alignment (genome) <i class="fas fa-toolbox"></i> [STAR](https://github.com/alexdobin/STAR), [HiSat2](https://ccb.jhu.edu/software/hisat2/index.shtml), [GSNAP](http://research-pub.gene.com/gmap/), [Novoalign](http://www.novocraft.com/products/novoalign/) (Commercial) .citation[ .cite[<i class="fas fa-link"></i> Baruzzo, Giacomo, *et al*. "Simulation-based comprehensive benchmarking of RNA-seq aligners." [Nature methods 14.2 (2017): 135](https://www.nature.com/articles/nmeth.4106)] ] --- name: aligner-speed ## Aligners • Speed .pull-left-50[  ] .pull-right-50[ |Program|Time_Min|Memory_GB| |-------|--------|---------| |HISATx1|22.7|4.3| |HISATx2|47.7|4.3| |HISAT|26.7|4.3| |STAR|25|28| |STARx2|50.5|28| |GSNAP|291.9|20.2| |TopHat2|1170|4.3| ] .citation[ .cite[<i class="fas fa-link"></i> Baruzzo, Giacomo, *et al*. "Simulation-based comprehensive benchmarking of RNA-seq aligners." [Nature methods 14.2 (2017): 135](https://www.nature.com/articles/nmeth.4106)] ] --- name: aligner-accuracy ## Aligners • Accuracy .size-70[] .size-40[] <i class="fas fa-toolbox"></i> [STAR](https://github.com/alexdobin/STAR), [HiSat2](https://ccb.jhu.edu/software/hisat2/index.shtml), [GSNAP](http://research-pub.gene.com/gmap/), [Novoalign](http://www.novocraft.com/products/novoalign/) (Commercial) .citation[ .cite[<i class="fas fa-link"></i> Baruzzo, Giacomo, *et al*. "Simulation-based comprehensive benchmarking of RNA-seq aligners." [Nature methods 14.2 (2017): 135](https://www.nature.com/articles/nmeth.4106)] ] --- name: mapping-required ## Mapping - Reads (FASTQ) ``` @ST-E00274:179:HHYMLALXX:8:1101:1641:1309 1:N:0:NGATGT NCATCGTGGTATTTGCACATCTTTTCTTATCAAATAAAAAGTTTAACCTACTCAGTTATGCGCATACGTTTTTTGATGGCATTTCCATAAACCGATTTTTTTTTTATGCACGTACCCAAAACGTGCAGAAAAATACGCTGCTAGAAATGTA + #AAAFAFA<-AFFJJJAFA-FFJJJJFFFAJJJJ-<FFJJJ-A-F-7--FA7F7-----FFFJFA<FFFFJ<AJ--FF-A<A-<JJ-7-7-<FF-FFFJAFFAA--A--7FJ-7----77-A--7F7)---7F-A----7)7-----7<<- ``` `@instrument:runid:flowcellid:lane:tile:xpos:ypos read:isfiltered:controlnumber:sampleid` - Reference Genome/Transcriptome (FASTA) ``` >1 dna:chromosome chromosome:GRCz10:1:1:58871917:1 REF GATCTTAAACATTTATTCCCCCTGCAAACATTTTCAATCATTACATTGTCATTTCCCCTC CAAATTAAATTTAGCCAGAGGCGCACAACATACGACCTCTAAAAAAGGTGCTGTAACATG ``` - Annotation (GTF/GFF) ``` #!genome-build GRCz10 #!genebuild-last-updated 2016-11 4 ensembl_havana gene 6732 52059 . - . gene_id "ENSDARG00000104632"; gene_version "2"; gene_name "rerg"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; havana_gene "OTTDARG00000044080"; havana_gene_version "1"; ``` `seq source feature start end score strand frame attribute` .citation[ .cite[<i class="fas fa-link"></i> Illumina read name [format](http://support.illumina.com/content/dam/illumina-support/help/BaseSpaceHelp_v2/Content/Vault/Informatics/Sequencing_Analysis/BS/swSEQ_mBS_FASTQFiles.htm), GTF [format](https://www.ensembl.org/info/website/upload/gff.html)] ] --- name: alignment ## Alignment - SAM/BAM (Sequence Alignment Map format) ``` ST-E00274:188:H3JWNCCXY:4:1102:32431:49900 163 1 1 60 8S139M4S = 385 535 TATTTAGAGATCTTAAACATCCATTCCCCCTGCAAACATTTTCAATCATTACATTGTCATTTTCCCTCCAAATTAAATTTAGCCAGAGGCGCACAACATACGACCTCTAAAAAAGGTGCTGGAACATGTACCTATATGCAGCACCACCATC AAAFAFFAFFFFJ7FFFFJ<JAFA7F-<AJ7JJ<FFFJ--<FAJF<7<7FAFJ-<AFA<-JJJ-AF-AJ-FF<F--A<FF<-7777-7JA-77A---F-7AAFF-FJA--77FJ<--77)))7<JJA<J77<-------<7--))7)))7- NM:i:4 MD:Z:12T0T40C58T25 AS:i:119 XS:i:102 XA:Z:17,-53287490,4S33M4D114M,11; MQ:i:60 MC:Z:151M RG:Z:ST-E00274_188_H3JWNCCXY_4 ``` `query flag ref pos mapq cigar mrnm mpos tlen seq qual opt` **Never store alignment files in raw SAM format. Always compress it!** |Format|Size_GB| |------|-------| |SAM|7.4| |BAM|1.9| |CRAM lossless Q|1.4| |CRAM 8 bins Q|0.8| |CRAM no Q|0.26| .citation[ .cite[<i class="fas fa-link"></i> SAM file [format](http://www.htslib.org/doc/sam.html)] ] --- name: vis-tview ## Visualisation • `tview` `samtools tview alignment.bam genome.fasta`  --- name: vis-igv ## Visualisation • IGV  <i class="fas fa-toolbox"></i> [IGV](http://software.broadinstitute.org/software/igv/), [UCSC Genome Browser](https://genome.ucsc.edu/) --- name: vis-seqmonk ## Visualisation • SeqMonk  <i class="fas fa-toolbox"></i> [SeqMonk](https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/) --- name: alignment-qc class: spaced ## Alignment QC - Number of reads mapped/unmapped/paired etc - Uniquely mapped - Insert size distribution - Coverage - Gene body coverage - Biotype counts / Chromosome counts - Counts by region: gene/intron/non-genic - Sequencing saturation - Strand specificity <i class="fas fa-toolbox"></i> STAR (final log file), samtools > stats, bamtools > stats, [QoRTs](https://hartleys.github.io/QoRTs/), [RSeQC](http://rseqc.sourceforge.net/), [Qualimap](http://qualimap.bioinfo.cipf.es/) --- name: star-log ## Alignment QC • STAR Log MultiQC can be used to summarise and plot STAR log files. .size-95[] --- name: qorts-region ## Alignment QC • Features QoRTs was run on all samples and summarised using MultiQC. .size-95[] --- name: qorts-plots ## Alignment QC • QoRTs .size-95[] --- name: alignment-qc-1 ## Alignment QC • Examples .pull-left-50[ **Soft clipping**  ] .pull-right-50[ **Gene body coverage**  ] --- name: alignment-qc-2 ## Alignment QC • Examples .pull-left-50[ **Insert size**  ] .pull-right-50[ **Saturation curve**  ] --- name: quantification-counts class: spaced ## Quantification • Counts .pull-left-50[ - Read counts = gene expression - Reads can be quantified on any feature (gene, transcript, exon etc) - Intersection on gene models - Gene/Transcript level  <i class="fas fa-toolbox"></i> [featureCounts](http://bioinf.wehi.edu.au/featureCounts/), [HTSeq](https://github.com/simon-anders/htseq) ] .pull-right-50[ .size-85[.center[]] ] --- name: quant-pcr-multi ## Quantification .pull-left-50[ **PCR duplicates** - Ignore for RNA-Seq data - Computational deduplication (Don't!) - Use PCR-free library-prep kits - Use UMIs during library-prep **Multi-mapping** - Added (BEDTools multicov) - Discard (featureCounts, HTSeq) - Distribute counts (Cufflinks, featureCounts) - Rescue - Probabilistic assignment (Rcount, Cufflinks) - Prioritise features (Rcount) - Probabilistic assignment with EM (RSEM) ] .pull-right-50[  ] .citation[ .cite[<i class="fas fa-link"></i> Fu, Yu, *et al*. "Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers." [BMC genomics 19.1 (2018): 531](https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-4933-1)] .cite[<i class="fas fa-link"></i> Parekh, Swati, *et al*. "The impact of amplification on differential expression analyses by RNA-seq." [Scientific reports 6 (2016): 25533](https://www.nature.com/articles/srep25533)] .cite[<i class="fas fa-link"></i> Klepikova, Anna V., *et al*. "Effect of method of deduplication on estimation of differential gene expression using RNA-seq." [PeerJ 5 (2017): e3091](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5357343/)] ] --- name: quantification-abundance ## Quantification • Abundance - Count methods - Provide no inference on isoforms - Cannot accurately measure fold change <!--.size-60[]--> - Probabilistic assignment - Deconvolute ambiguous mappings - Transcript-level - cDNA reference **Kallisto, Salmon** - Ultra-fast & alignment-free - Subsampling & quantification confidence - Transcript-level estimates improves gene-level estimates - Kallisto/Salmon > transcript-counts > `tximport()` > gene-counts <i class="fas fa-toolbox"></i> [RSEM](https://deweylab.github.io/RSEM/), [Kallisto](https://pachterlab.github.io/kallisto/), [Salmon](https://combine-lab.github.io/salmon/), [Cufflinks2](http://cole-trapnell-lab.github.io/cufflinks/) .citation[ .cite[<i class="fas fa-link"></i> Soneson, Charlotte, *et al*. "Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences." [F1000Research 4 (2015)](https://f1000research.com/articles/4-1521/v2)] .cite[<i class="fas fa-link"></i> Zhang, Chi, *et al*. "Evaluation and comparison of computational tools for RNA-seq isoform quantification." [BMC genomics 18.1 (2017): 583](https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-4002-1)] ] --- name: quantification-qc ## Quantification QC ``` ENSG00000000003 140 242 188 143 287 344 438 280 253 ENSG00000000005 0 0 0 0 0 0 0 0 0 ENSG00000000419 69 98 77 55 52 94 116 79 69 ENSG00000000457 56 75 104 79 157 205 183 178 153 ENSG00000000460 33 27 23 19 27 42 69 44 40 ENSG00000000938 7 38 13 17 35 76 53 37 24 ENSG00000000971 545 878 694 636 647 216 492 798 323 ENSG00000001036 79 154 74 80 128 167 220 147 72 ``` .pull-left-50[ - Pairwise correlation between samples must be high (>0.9) .size-60[] ] .pull-right-50[ - Count QC using RNASeqComp .size-80[] ] <i class="fas fa-toolbox"></i> [RNASeqComp](https://bioconductor.org/packages/release/bioc/html/rnaseqcomp.html) .citation[ .cite[<i class="fas fa-link"></i> Teng, Mingxiang, *et al*. "A benchmark for RNA-seq quantification pipelines." [Genome biology 17.1 (2016): 74](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0940-1)] ] --- name: multiqc ## MultiQC [](https://multiqc.info/examples/rna-seq/multiqc_report.html) --- name: normalisation ## Normalisation - Control for Sequencing depth & compositional bias - Median of Ratios (DESeq2) and TMM (edgeR) perform the best  - For DGE using DGE packages, use raw counts - For clustering, heatmaps etc use VST, VOOM or RLOG - For own analysis, plots etc, use TPM - Other solutions: spike-ins/house-keeping genes <img src="data/rnaseq/distribution.jpg" style="height:240px; position:fixed; right:0px; bottom: 0px; margin-right: 70px; margin-bottom: 140px;"> .citation[ .cite[<i class="fas fa-link"></i> Dillies, Marie-Agnes, *et al*. "A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis." [Briefings in bioinformatics 14.6 (2013): 671-683](https://www.ncbi.nlm.nih.gov/pubmed/22988256)] .cite[<i class="fas fa-link"></i> Evans, Ciaran, Johanna Hardin, and Daniel M. Stoebel. "Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions." [Briefings in bioinformatics (2017)](https://arxiv.org/abs/1609.00959)] .cite[<i class="fas fa-link"></i> Wagner, Gunter P., Koryu Kin, and Vincent J. Lynch. "Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples." [Theory in biosciences 131.4 (2012): 281-285](https://link.springer.com/article/10.1007/s12064-012-0162-3)] ] --- name: exploratory-heatmap ## Exploratory • Heatmap - Remove lowly expressed genes - Transform raw counts to VST, VOOM, RLOG, TPM etc - Sample-sample clustering heatmap .size-70[] <i class="fas fa-toolbox"></i> [`pheatmap()`](https://github.com/raivokolde/pheatmap) --- name: exploratory-pca ## Exploratory • PCA .size-90[] --- name: batch-correction class: spaced ## Batch correction - Estimate variation explained by variables (PVCA) .size-70[] - Find confounding effects as surrogate variables (SVA) - Model known batches in the LM/GLM model - Correct known batches (ComBat)(Harsh!) - Interactively evaluate batch effects and correction (BatchQC) <i class="fas fa-toolbox"></i> [SVA](http://bioconductor.org/packages/release/bioc/html/sva.html), [PVCA](https://bioconductor.org/packages/release/bioc/html/pvca.html), [BatchQC](http://bioconductor.org/packages/release/bioc/html/BatchQC.html) .citation[ .cite[<i class="fas fa-link"></i> Liu, Qian, and Marianthi Markatou. "Evaluation of methods in removing batch effects on RNA-seq data." [Infectious Diseases and Translational Medicine 2.1 (2016): 3-9](http://www.tran-med.com/article/2016/2411-2917-2-1-3.html)] .cite[<i class="fas fa-link"></i> Manimaran, Solaiappan, et al. "BatchQC: interactive software for evaluating sample and batch effects in genomic data." [Bioinformatics 32.24 (2016): 3836-3838](https://academic.oup.com/bioinformatics/article/32/24/3836/2525651)] ] --- name: dge-1 ## DGE .size-90[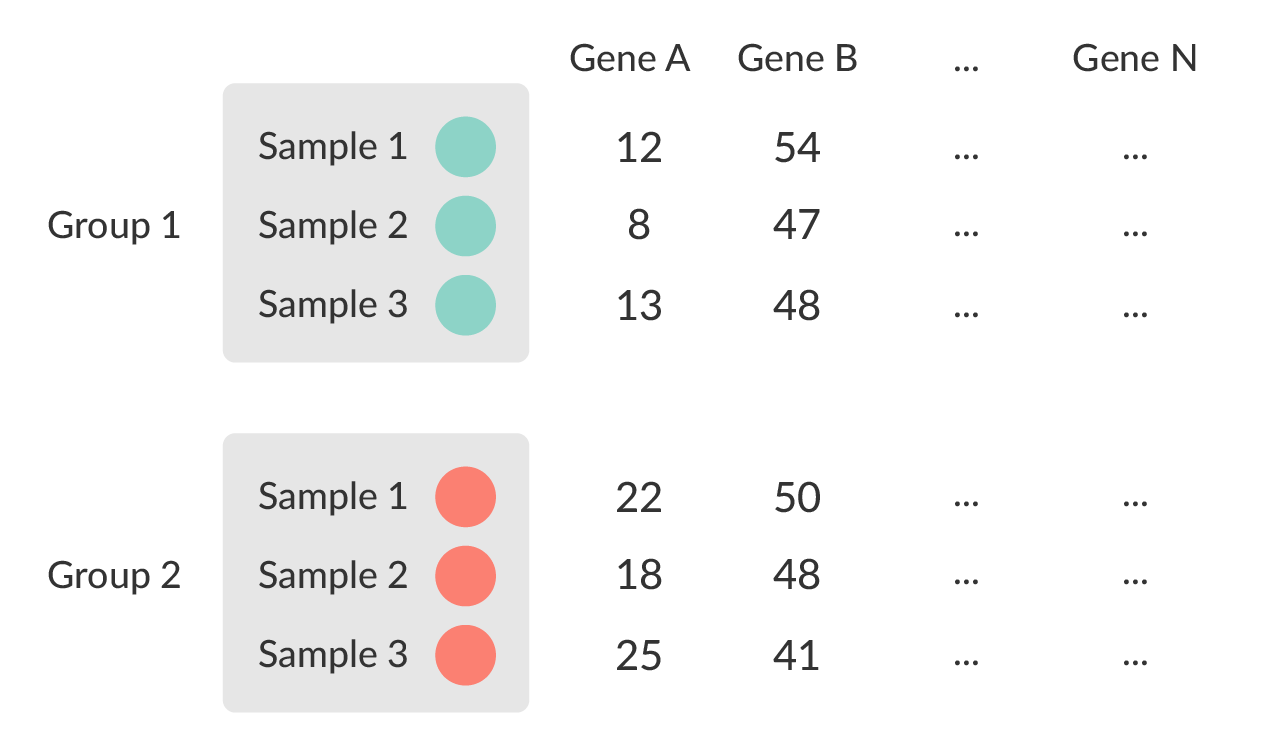] --- name: dge-2 ## DGE - DESeq2, edgeR (Neg-binom > GLM > Test), Limma-Voom (Neg-binom > Voom-transform > LM > Test) - DESeq2 `~age+condition` - Estimate size factors `estimateSizeFactors()` - Estimate gene-wise dispersion `estimateDispersions()` - Fit curve to gene-wise dispersion estimates - Shrink gene-wise dispersion estimates - GLM fit for each gene - Wald test `nbinomWaldTest()` .size-50[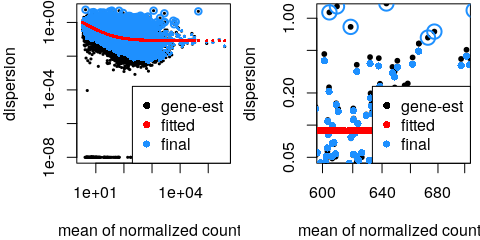] <i class="fas fa-toolbox"></i> [DESeq2](), [edgeR](), [Limma-Voom]() .citation[ .cite[<i class="fas fa-link"></i> Seyednasrollah, Fatemeh, *et al*. "Comparison of software packages for detecting differential expression in RNA-seq studies." [Briefings in bioinformatics 16.1 (2013): 59-70](https://academic.oup.com/bib/article/16/1/59/240754)] ] --- name: dge-3 ## DGE - Results `results()` ``` log2 fold change (MLE): type type2 vs control Wald test p-value: type type2 vs control DataFrame with 1 row and 6 columns baseMean log2FoldChange lfcSE <numeric> <numeric> <numeric> ENSG00000000003 242.307796723287 -0.932926089608546 0.114285150312285 stat pvalue padj <numeric> <numeric> <numeric> ENSG00000000003 -8.16314356729037 3.26416150242775e-16 1.36240609998527e-14 ``` - Summary `summary()` ``` out of 17889 with nonzero total read count adjusted p-value < 0.1 LFC > 0 (up) : 4526, 25% LFC < 0 (down) : 5062, 28% outliers [1] : 25, 0.14% low counts [2] : 0, 0% (mean count < 3) [1] see 'cooksCutoff' argument of ?results [2] see 'independentFiltering' argument of ?results ``` --- name: dge-4 ## DGE .pull-left-50[ - MA plot `plotMA()` 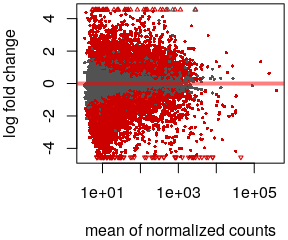 - Volcano plot 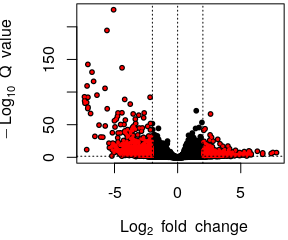 ] .pull-right-50[ - Normalised counts `plotCounts()`  <img src="data/rnaseq/scattered.gif" style="height:220px; position:fixed; right:0px; bottom:0px; margin-right: 130px; margin-bottom: 10px;"> ] --- name: functional-analysis-1 ## Functional analysis • GO - Gene set analysis (GSA) - Gene set enrichment analysis (GSEA) - Gene ontology / Reactome databases .size-70[] <img src="data/rnaseq/systembio.png" style="height:220px; position:fixed; right:0px; bottom:0px; margin-right: 70px; margin-bottom: 380px;"> --- name: functional-analysis-2 ## Functional analysis • Kegg - Pathway analysis (Kegg)  <i class="fas fa-toolbox"></i> [DAVID](https://david.ncifcrf.gov/), [clusterProfiler](https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html), [ClueGO](http://apps.cytoscape.org/apps/cluego), [ErmineJ](https://erminej.msl.ubc.ca/), [pathview](https://bioconductor.org/packages/release/bioc/html/pathview.html) --- name: summary class: spaced ## Summary - Sound experimental design to avoid confounding - Plan carefully about lib prep, sequencing etc based on experimental objective - Biological replicates may be more important than paired-end reads or long reads - Discard low quality bases, reads, genes and samples - Verify that tools and methods align with data assumptions - Experiment with multiple pipelines and tools - QC! QC everything at every step .large[<i class="fas fa-link"></i> Conesa, Ana, *et al.* "A survey of best practices for RNA-seq data analysis." [Genome biology 17.1 (2016): 13](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8)] --- name: help class: spaced ## Further learning - Griffith lab [RNA-Seq using HiSat & StringTie tutorial](https://github.com/griffithlab/rnaseq_tutorial/wiki) - SciLifeLab [courses](https://www.scilifelab.se/education/courses%26training) - HBC Training [DGE using DeSeq2 tutorial](https://github.com/hbctraining/Intro-to-R-with-DGE) - Hemberg lab [scRNA-Seq tutorial](http://hemberg-lab.github.io/scRNA.seq.course/index.html) - [RNA-Seq Blog](https://www.rna-seqblog.com/) <img src="data/rnaseq/help.png" style="height:200px; position:fixed; right:0px; bottom:0px; margin-right: 100px; margin-bottom: 20px;"> --- name: end_slide class: end-slide, middle count: false # Thank you. Questions? .end-text[ <p>R version 4.0.5 (2021-03-31)<br><p>Platform: x86_64-pc-linux-gnu (64-bit)</p><p>OS: Ubuntu 18.04.5 LTS</p><br> <span class="small">Built on : <i class='fa fa-calendar' aria-hidden='true'></i> 25-May-2021 at <i class='fa fa-clock-o' aria-hidden='true'></i> 13:35:25</span> <b>2021</b> • [SciLifeLab](https://www.scilifelab.se/) • [NBIS](https://nbis.se/) ] --- name: lab1 class: spaced ## Hands-On tutorial **Main exercise** - 01 Check the quality of the raw reads with **FastQC** - 02 Map the reads to the reference genome using **HISAT2** - 03 Assess the post-alignment quality using **QualiMap** - 04 Count the reads overlapping with genes using **featureCounts** - 05 Find DE genes using **DESeq2** in R **Bonus exercises** - 01 Functional annotation of DE genes using **GO/Reactome/Kegg** databases - 02 RNA-Seq figures and plots using **R** - 03 Visualisation of RNA-seq BAM files using **IGV** genome browser .large[**Data:** `/sw/courses/ngsintro/rnaseq/`] .large[**Work:** `/proj/gXXXXXXX/nobackup/<user>/rnaseq/`] --- name: lab2 ## Hands-On tutorial .pull-left-50[ - Course data directory .large[`/sw/courses/ngsintro/rnaseq/`] .large[ ``` rnaseq/ +-- bonus/ | +-- assembly/ | +-- exon/ | +-- funannot/ | +-- plots/ +-- documents/ +-- main/ +-- 1_raw/ +-- 2_fastqc/ +-- 3_mapping/ +-- 4_qualimap/ +-- 5_dge/ +-- 6_multiqc/ +-- reference/ | +-- mouse_chr19_hisat2/ +-- scripts/ ``` ] ] .pull-right-50[ - Your work directory .large[`/proj/gXXXX/nobackup/<user>/`] .large[ ``` [user]/ rnaseq/ +-- 1_raw/ +-- 2_fastqc/ +-- 3_mapping/ +-- 4_qualimap/ +-- 5_dge/ +-- 6_multiqc/ +-- reference/ | +-- mouse_chr19_hisat2/ +-- scripts/ +-- funannot/ +-- plots/ ``` ] ]