Differential gene expression¶
In this tutorial we will cover about Differetial gene expression, which comprises an extensive range of topics and methods. In single cell, differential expresison can have multiple functionalities such as of identifying marker genes for cell populations, as well as differentially regulated genes across conditions (healthy vs control). We will also exercise on how to account the batch information in your test.
Differential expression is performed with the function rank_genes_group. The default method to compute differential expression is the t-test_overestim_var. Other implemented methods are: logreg, t-test and wilcoxon.
By default, the .raw attribute of AnnData is used in case it has been initialized, it can be changed by setting use_raw=False.
The clustering with resolution 0.6 seems to give a reasonable number of clusters, so we will use that clustering for all DE tests.
First, let's import libraries and fetch the clustered data from the previous lab.
import numpy as np
import pandas as pd
import scanpy as sc
import gseapy
import matplotlib.pyplot as plt
sc.settings.verbosity = 2 # verbosity: errors (0), warnings (1), info (2), hints (3)
#sc.logging.print_versions()
sc.settings.set_figure_params(dpi=80)
Read in the clustered data object.
adata = sc.read_h5ad('./data/results/scanpy_clustered_covid.h5ad')
print(adata.X.shape)
print(adata.raw.X.shape)
print(adata.raw.X[:10,:10])
As you can see, the X matrix only contains the variable genes, while the raw matrix contains all genes.
Printing a few of the values in adata.raw.X shows that the raw matrix is not normalized.
For DGE analysis we would like to run with all genes, but on normalized values, so we will have to revert back to the raw matrix and renormalize.
adata = adata.raw.to_adata()
sc.pp.normalize_per_cell(adata, counts_per_cell_after=1e4)
sc.pp.log1p(adata)
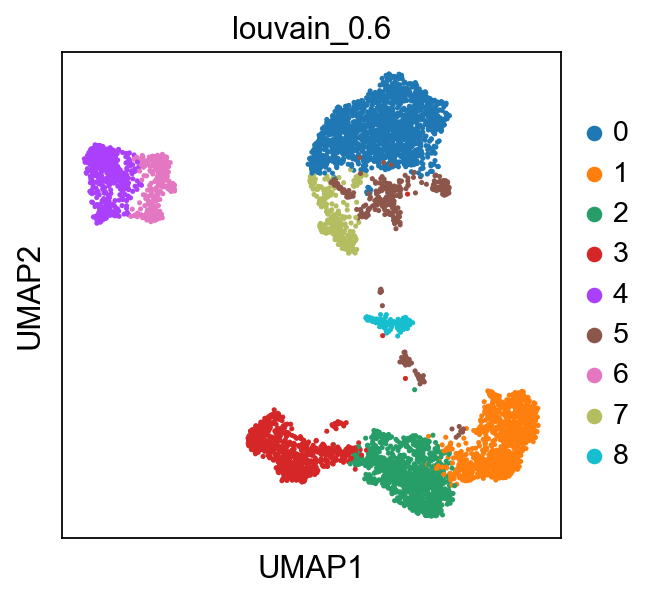
Now lets look at the clustering of the object we loaded in the umap. We will use louvain_0.6 clustering in this exercise.
sc.pl.umap(adata, color='louvain_0.6')
T-test¶
sc.tl.rank_genes_groups(adata, 'louvain_0.6', method='t-test', key_added = "t-test")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False, key = "t-test")
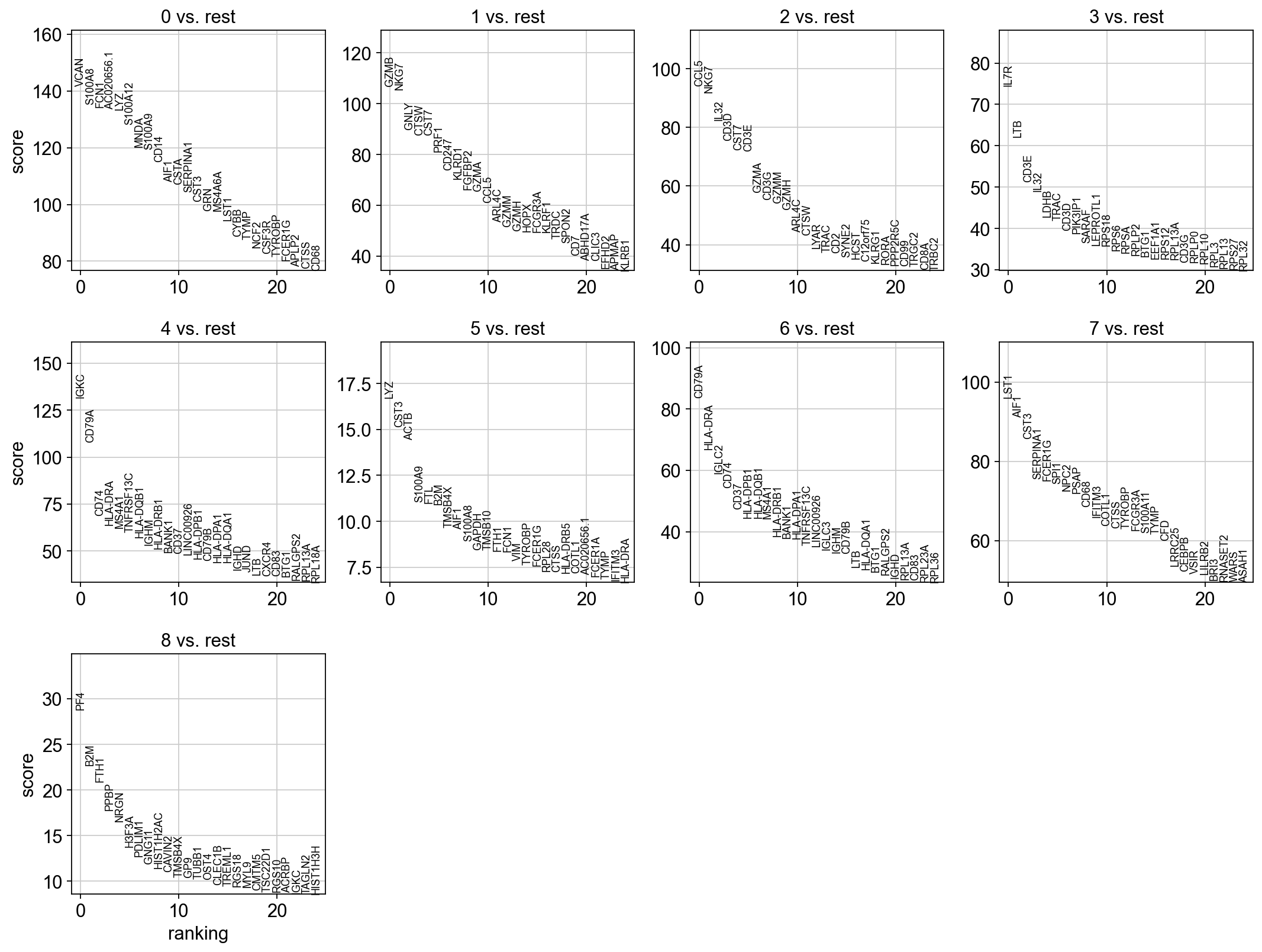
# results are stored in the adata.uns["t-test"] slot
adata
T-test overestimated_variance¶
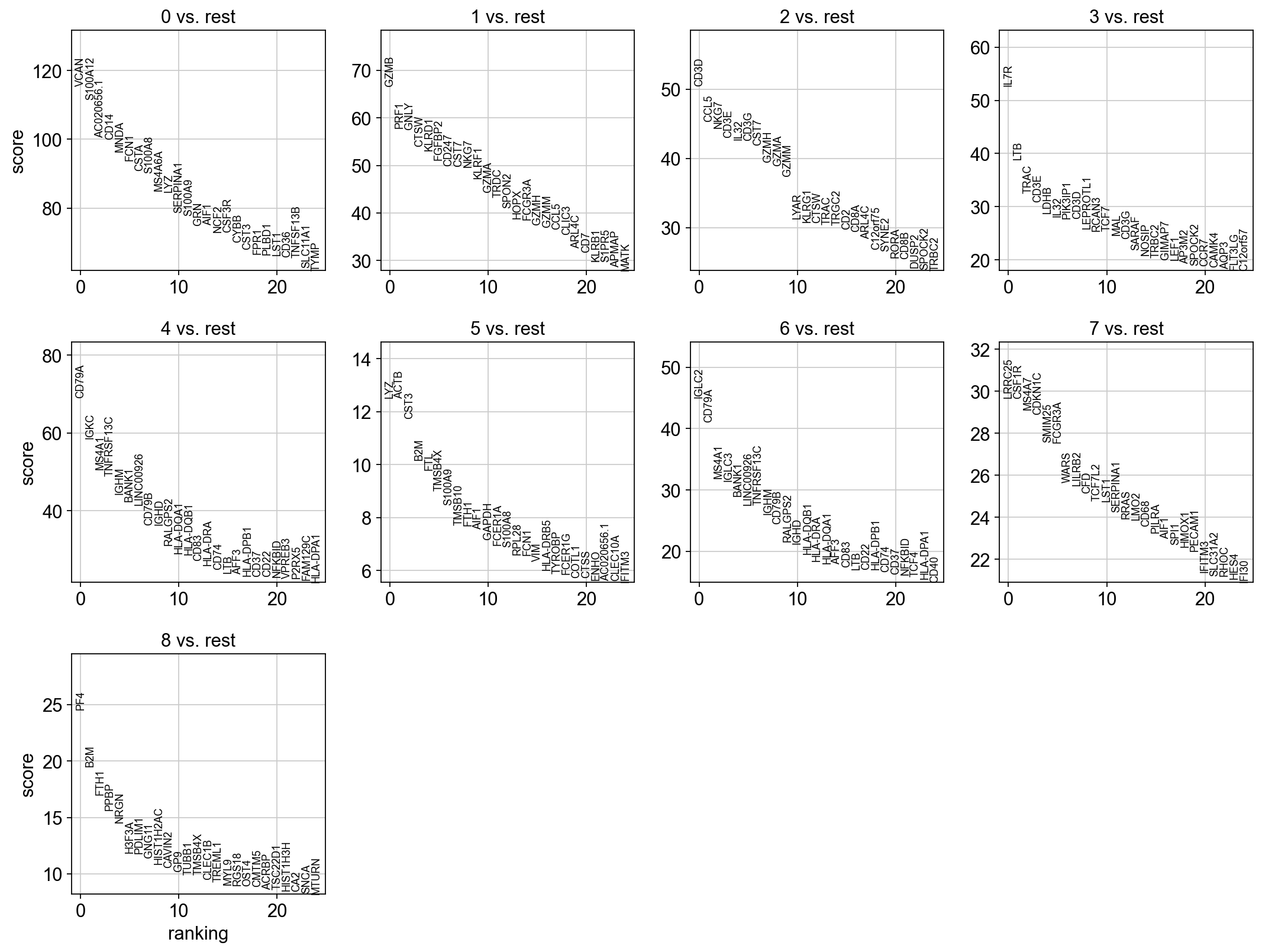
sc.tl.rank_genes_groups(adata, 'louvain_0.6', method='t-test_overestim_var', key_added = "t-test_ov")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False, key = "t-test_ov")
Wilcoxon rank-sum¶
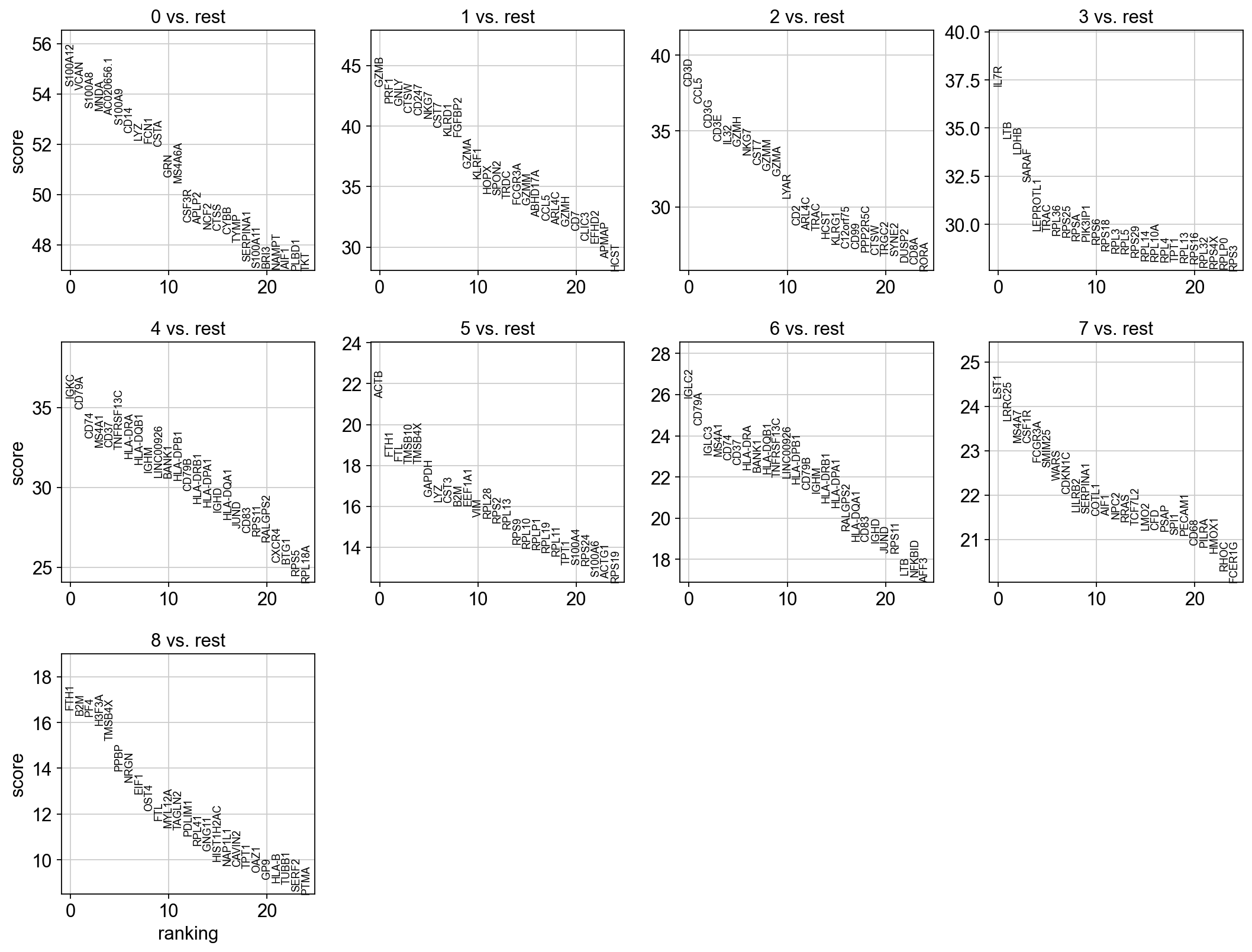
The result of a Wilcoxon rank-sum (Mann-Whitney-U) test is very similar. We recommend using the latter in publications, see e.g., Sonison & Robinson (2018). You might also consider much more powerful differential testing packages like MAST, limma, DESeq2 and, for python, the recent diffxpy.
sc.tl.rank_genes_groups(adata, 'louvain_0.6', method='wilcoxon', key_added = "wilcoxon")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False, key="wilcoxon")
Logistic regression test¶
As an alternative, let us rank genes using logistic regression. For instance, this has been suggested by Natranos et al. (2018). The essential difference is that here, we use a multi-variate appraoch whereas conventional differential tests are uni-variate. Clark et al. (2014) has more details.
sc.tl.rank_genes_groups(adata, 'louvain_0.6', method='logreg',key_added = "logreg")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False, key = "logreg")
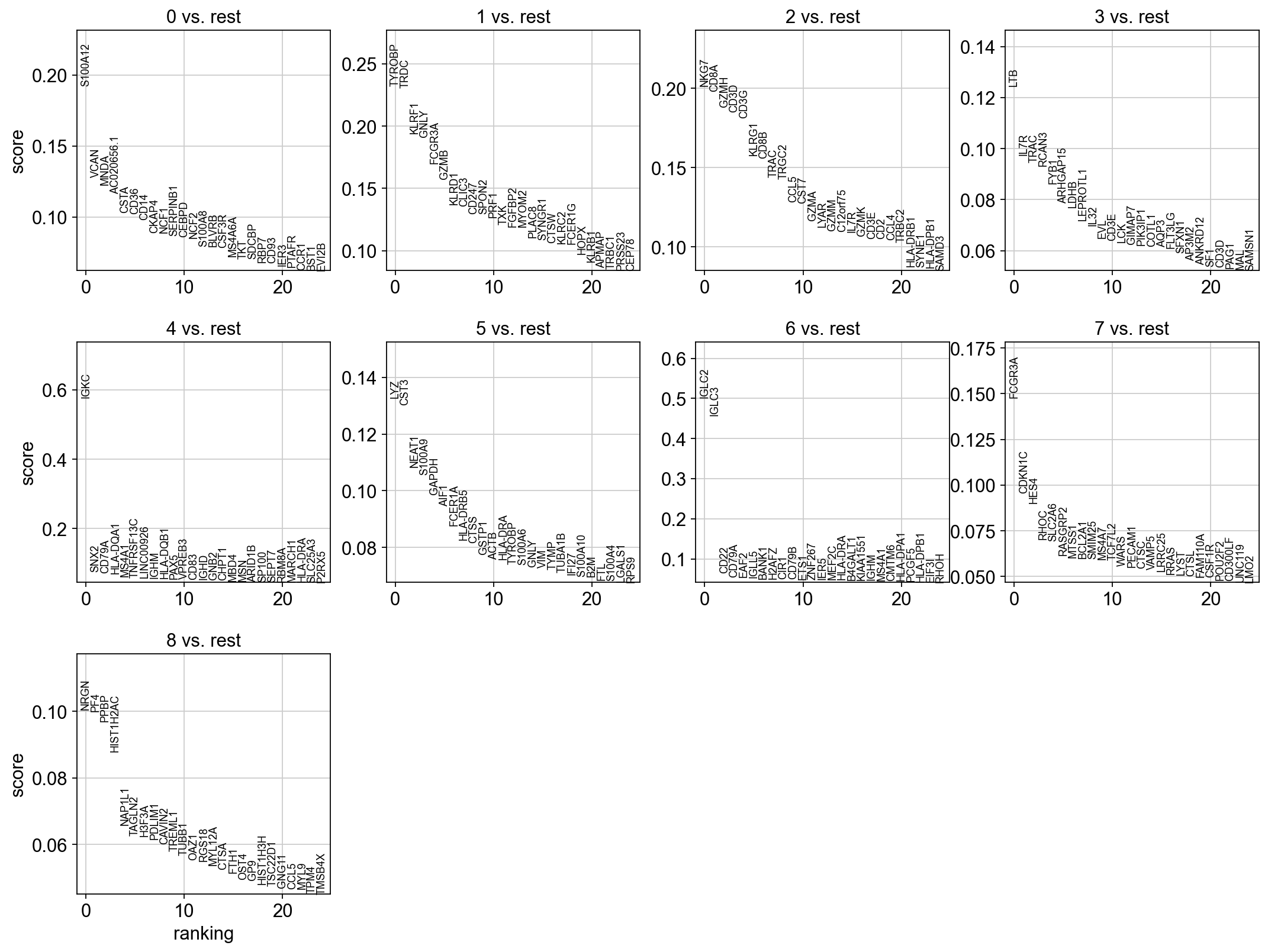
Compare genes¶
Take all significant DE genes for cluster0 with each test and compare the overlap.
#compare cluster1 genes, only stores top 100 by default
wc = sc.get.rank_genes_groups_df(adata, group='0', key='wilcoxon', pval_cutoff=0.01, log2fc_min=0)['names']
tt = sc.get.rank_genes_groups_df(adata, group='0', key='t-test', pval_cutoff=0.01, log2fc_min=0)['names']
tt_ov = sc.get.rank_genes_groups_df(adata, group='0', key='t-test_ov', pval_cutoff=0.01, log2fc_min=0)['names']
from matplotlib_venn import venn3
venn3([set(wc),set(tt),set(tt_ov)], ('Wilcox','T-test','T-test_ov') )
plt.show()
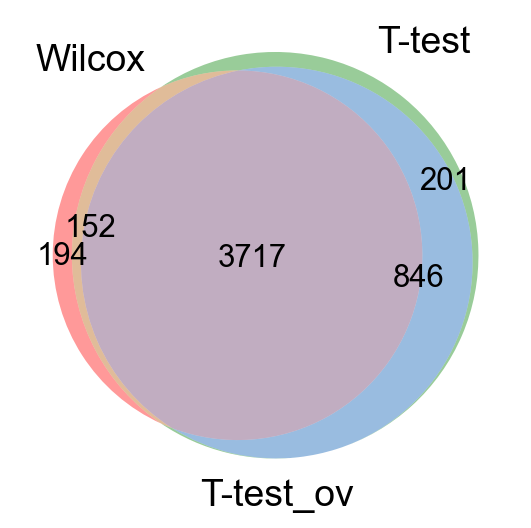
As you can see, the Wilcoxon test and the T-test with overestimated variance gives very similar result. Also the regular T-test has good overlap, while the Logistic regression gives quite different genes.
Visualization¶
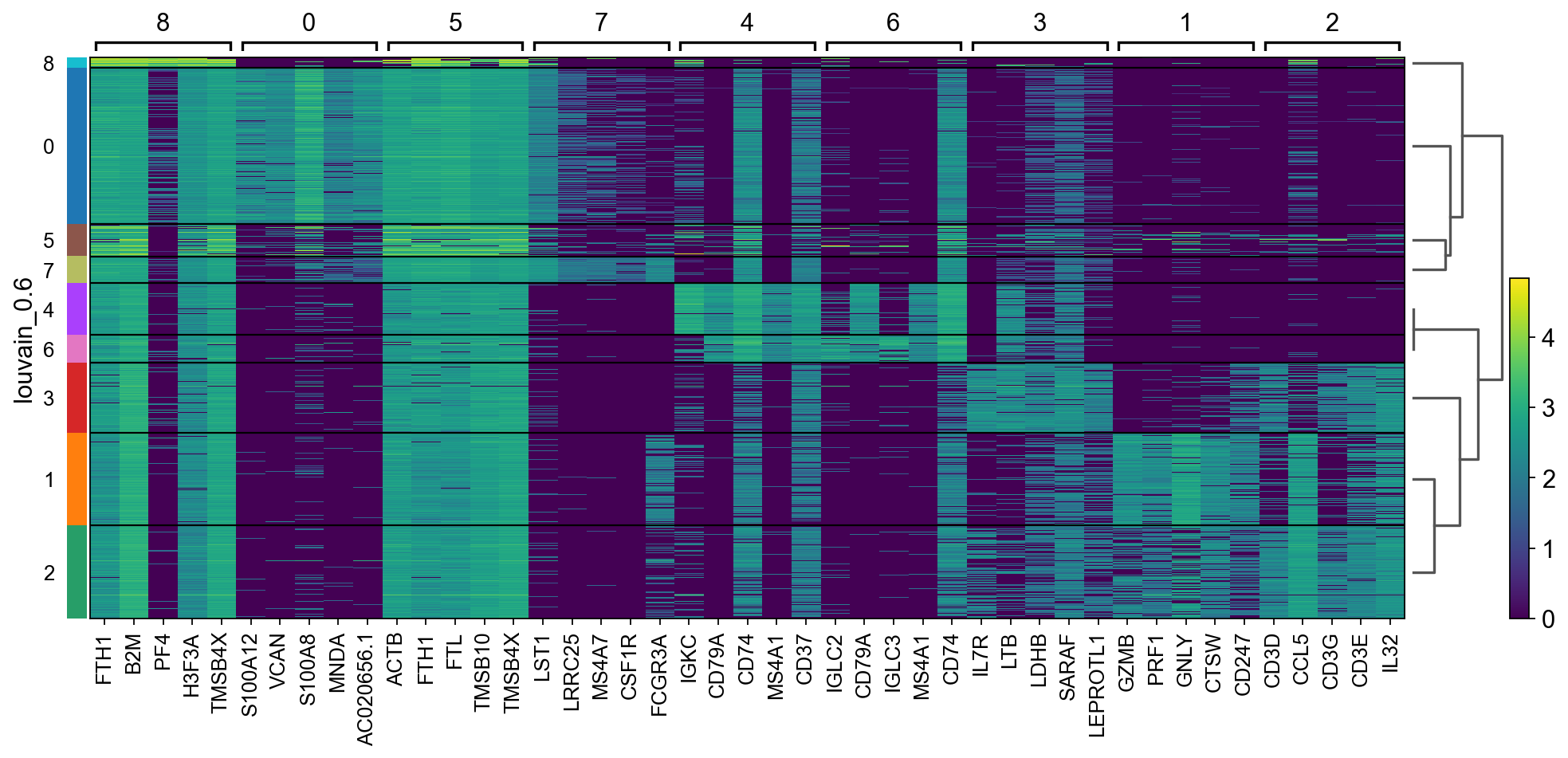
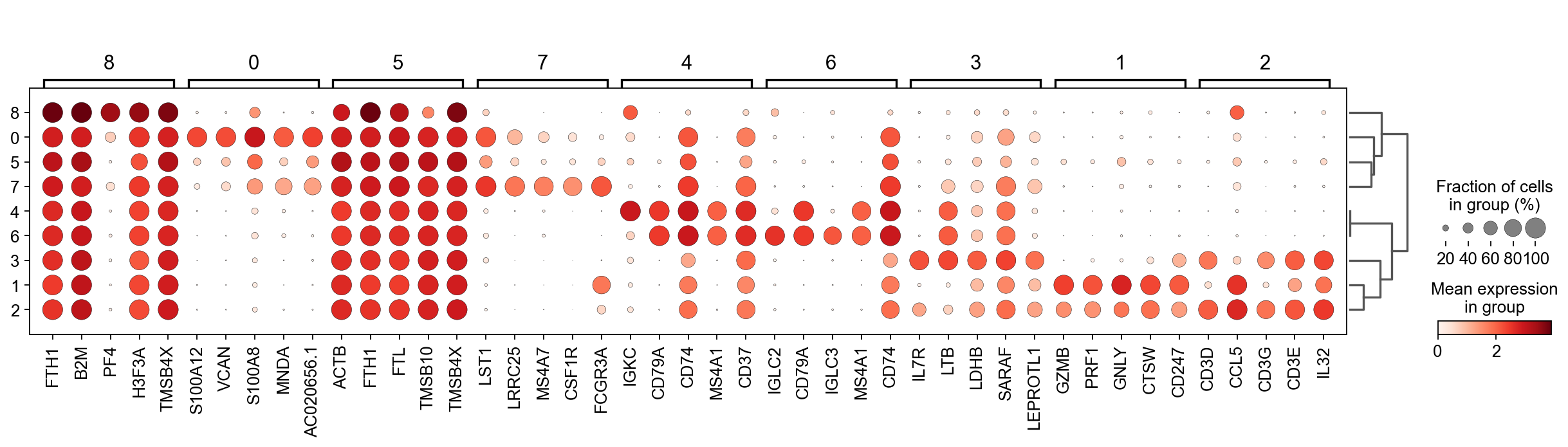
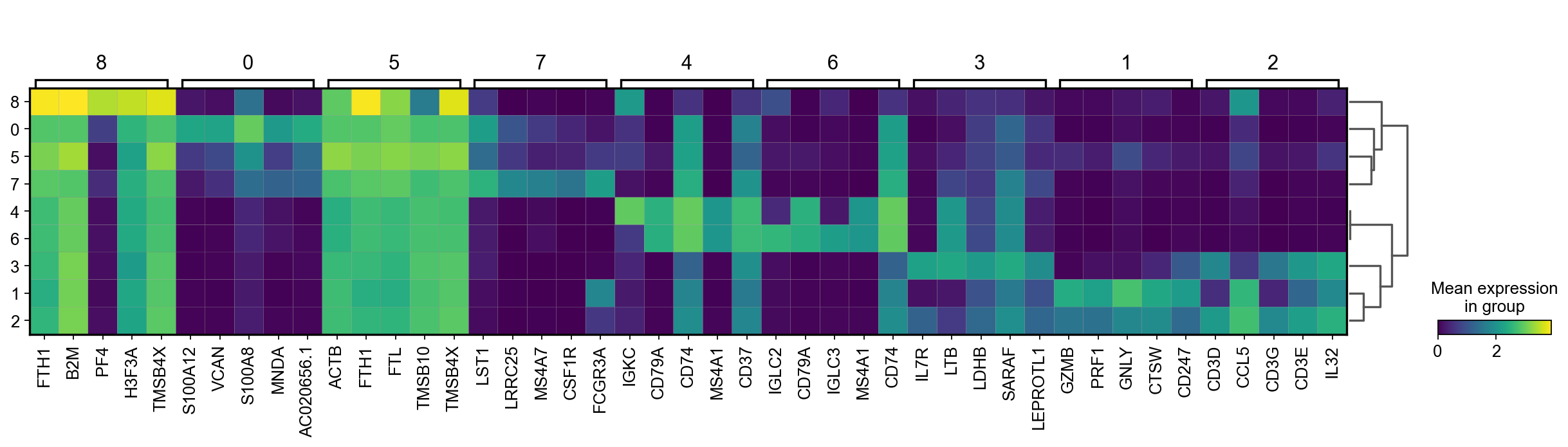
There are several ways to visualize the expression of top DE genes. Here we will plot top 5 genes per cluster from Wilcoxon test as heatmap, dotplot, violin plot or matrix.
sc.pl.rank_genes_groups_heatmap(adata, n_genes=5, key="wilcoxon", groupby="louvain_0.6", show_gene_labels=True)
sc.pl.rank_genes_groups_dotplot(adata, n_genes=5, key="wilcoxon", groupby="louvain_0.6")
sc.pl.rank_genes_groups_stacked_violin(adata, n_genes=5, key="wilcoxon", groupby="louvain_0.6")
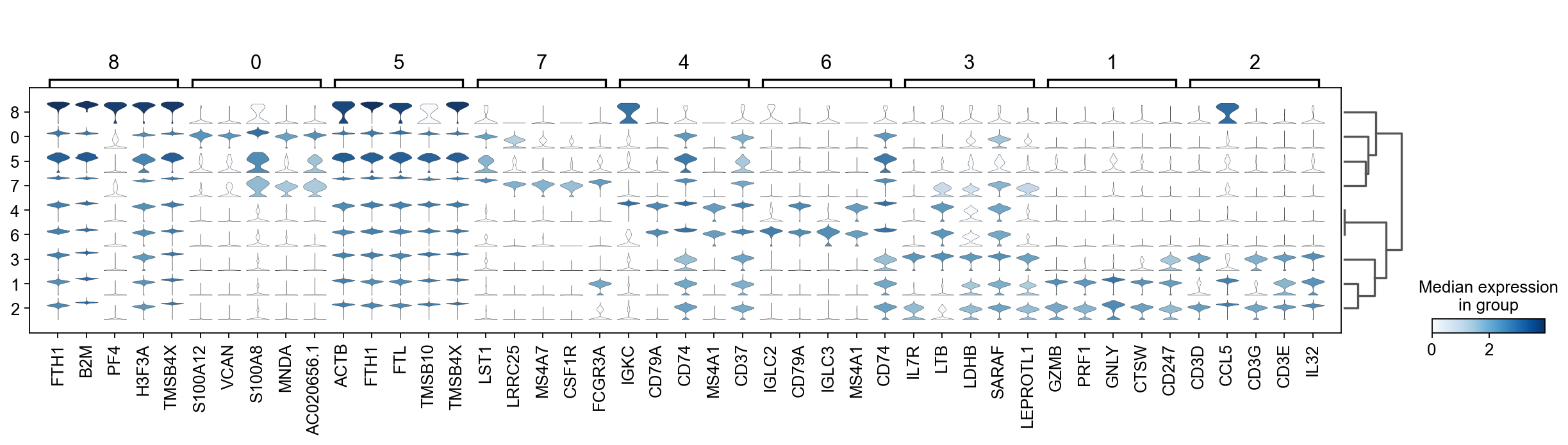
sc.pl.rank_genes_groups_matrixplot(adata, n_genes=5, key="wilcoxon", groupby="louvain_0.6")
Compare specific clusters¶
We can also do pairwise comparisons of individual clusters on one vs many clusters.
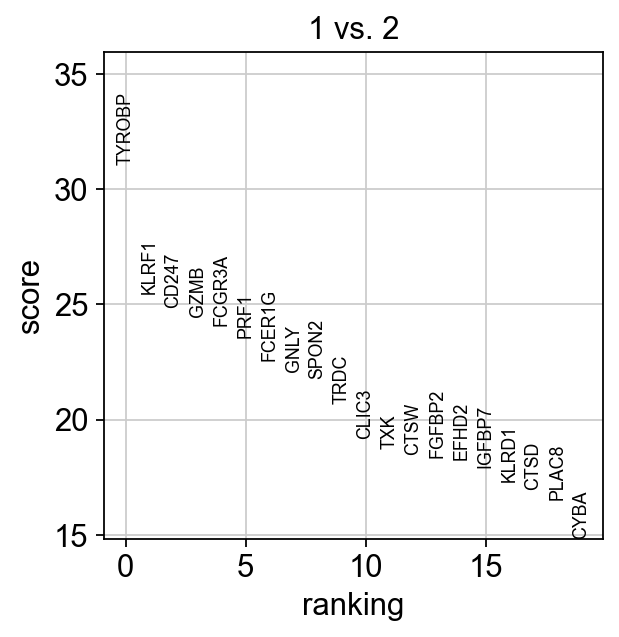
For instance, clusters 1 & 2 have very similar expression profiles.
sc.tl.rank_genes_groups(adata, 'louvain_0.6', groups=['1'], reference='2', method='wilcoxon')
sc.pl.rank_genes_groups(adata, groups=['1'], n_genes=20)
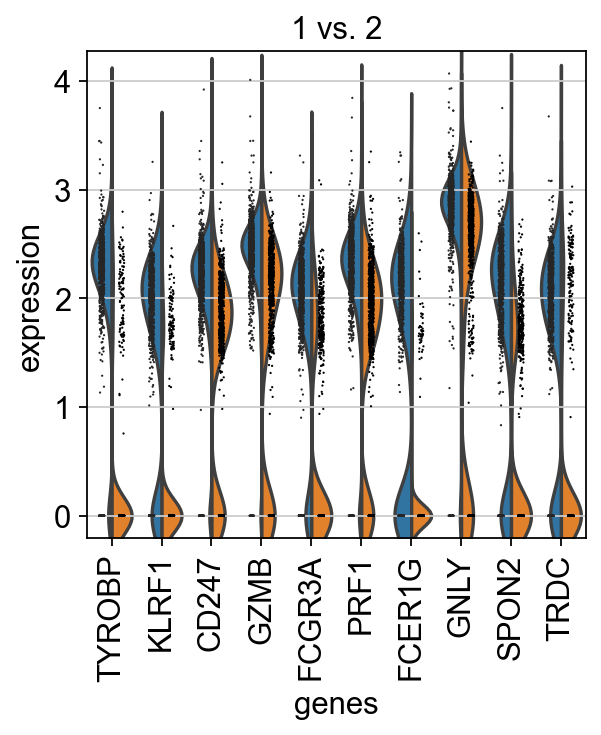
Plot as violins for those two groups.
sc.pl.rank_genes_groups_violin(adata, groups='1', n_genes=10)
# plot the same genes as violins across all the datasets.
# convert numpy.recarray to list
mynames = [x[0] for x in adata.uns['rank_genes_groups']['names'][:10]]
sc.pl.stacked_violin(adata, mynames, groupby = 'louvain_0.6')
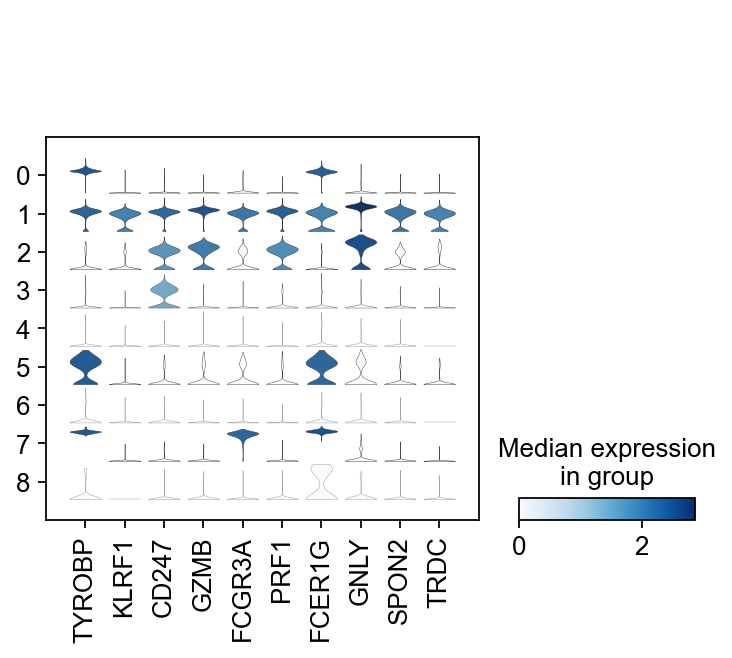
Differential expression across conditions¶
The second way of computing differential expression is to answer which genes are differentially expressed within a cluster. For example, in our case we have libraries comming from patients and controls and we would like to know which genes are influenced the most in a particular cell type.
For this end, we will first subset our data for the desired cell cluster, then change the cell identities to the variable of comparison (which now in our case is the "type", e.g. Covid/Ctrl).
cl1 = adata[adata.obs['louvain_0.6'] == '4',:]
cl1.obs['type'].value_counts()
sc.tl.rank_genes_groups(cl1, 'type', method='wilcoxon', key_added = "wilcoxon")
sc.pl.rank_genes_groups(cl1, n_genes=25, sharey=False, key="wilcoxon")
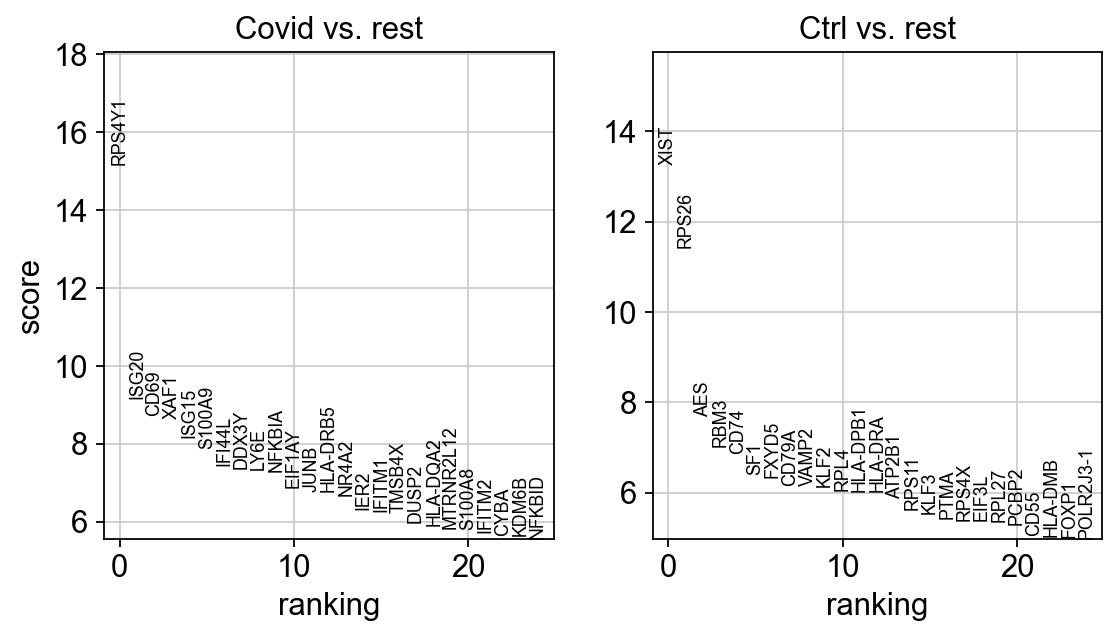
sc.pl.rank_genes_groups_violin(cl1, n_genes=10, key="wilcoxon")
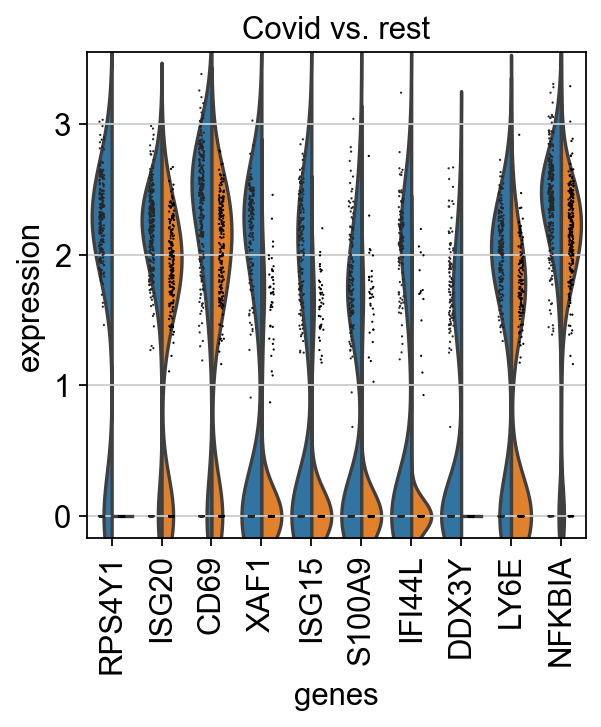
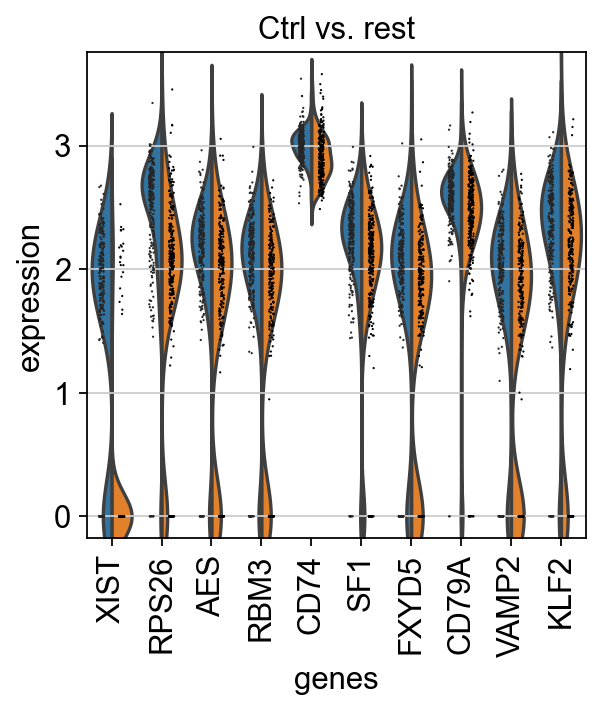
We can also plot these genes across all clusters, but split by "type", to check if the genes are also up/downregulated in other celltypes.
import seaborn as sns
genes1 = sc.get.rank_genes_groups_df(cl1, group='Covid', key='wilcoxon')['names'][:5]
genes2 = sc.get.rank_genes_groups_df(cl1, group='Ctrl', key='wilcoxon')['names'][:5]
genes = genes1.tolist() + genes2.tolist()
df = sc.get.obs_df(adata, genes + ['louvain_0.6','type'], use_raw=False)
df2 = df.melt(id_vars=["louvain_0.6",'type'], value_vars=genes)
sns.catplot(x = "louvain_0.6", y = "value", hue = "type", kind = 'violin',
col = "variable", data = df2, col_wrap=4, inner=None)
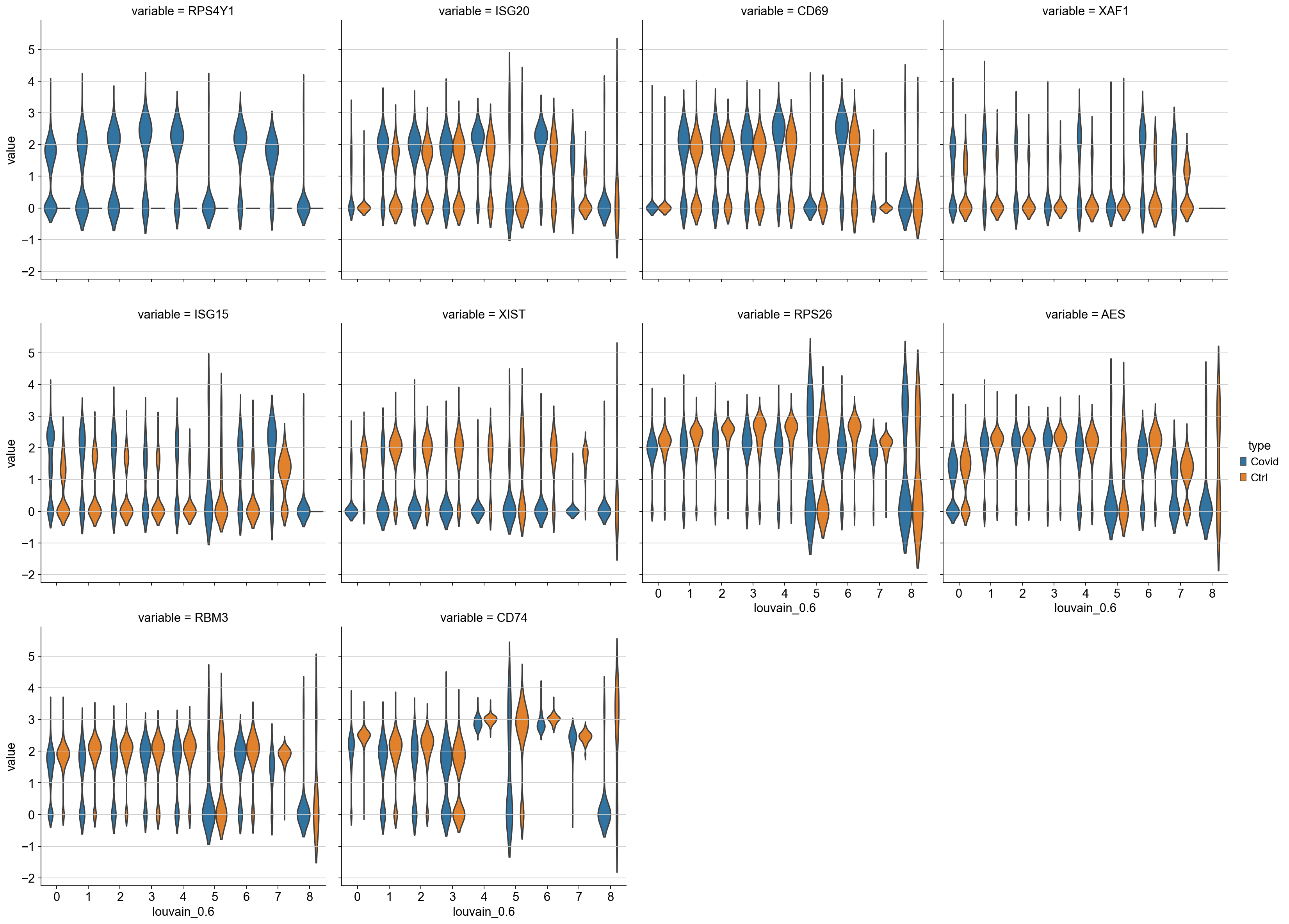
As you can see, we hwve many sex chromosome related genes among the top DE genes. And if you remember from the QC lab, we have inbalanced sex distribution among our subjects, so this may not be related to covid at all.
Remove sex chromosome genes¶
To remove some of the bias due to inbalanced sex in the subjects we can remove the sex chromosome related genes.
annot = sc.queries.biomart_annotations(
"hsapiens",
["ensembl_gene_id", "external_gene_name", "start_position", "end_position", "chromosome_name"],
).set_index("external_gene_name")
chrY_genes = adata.var_names.intersection(annot.index[annot.chromosome_name == "Y"])
chrX_genes = adata.var_names.intersection(annot.index[annot.chromosome_name == "X"])
sex_genes = chrY_genes.union(chrX_genes)
print(len(sex_genes))
all_genes = cl1.var.index.tolist()
print(len(all_genes))
keep_genes = [x for x in all_genes if x not in sex_genes]
print(len(keep_genes))
cl1 = cl1[:,keep_genes]
Rerun differential expression.
sc.tl.rank_genes_groups(cl1, 'type', method='wilcoxon', key_added = "wilcoxon")
sc.pl.rank_genes_groups(cl1, n_genes=25, sharey=False, key="wilcoxon")
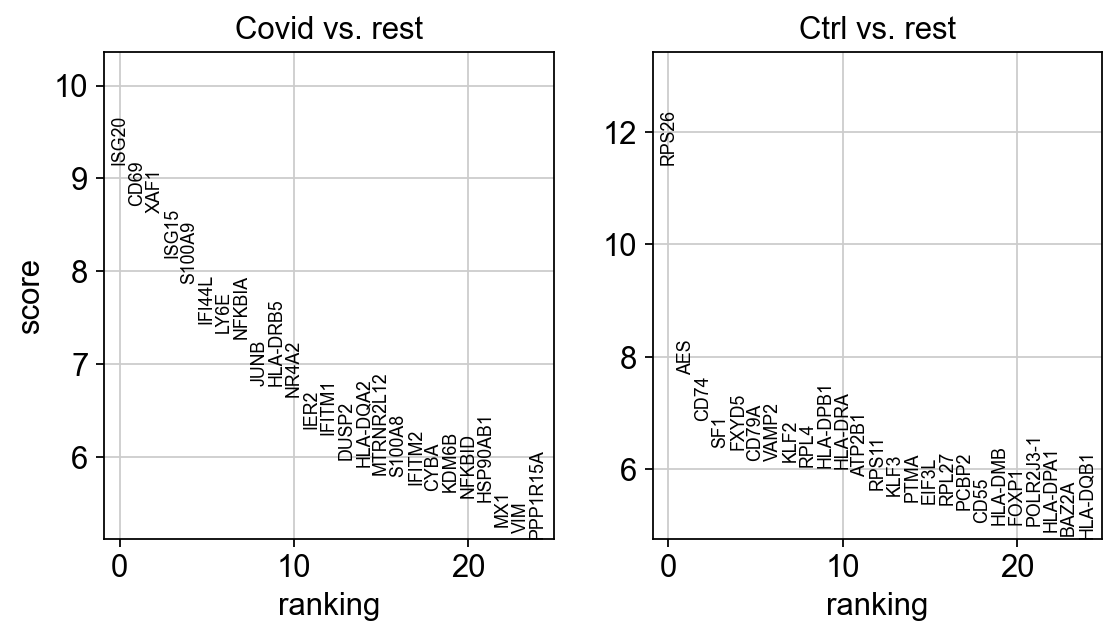
Patient Batch effects¶
When we are testing for Covid vs Control we are running a DGE test for 3 vs 3 individuals. That will be very sensitive to sample differences unless we find a way to control for it. So first, lets check how the top DGEs are expressed across the individuals:
genes1 = sc.get.rank_genes_groups_df(cl1, group='Covid', key='wilcoxon')['names'][:5]
genes2 = sc.get.rank_genes_groups_df(cl1, group='Ctrl', key='wilcoxon')['names'][:5]
genes = genes1.tolist() + genes2.tolist()
sc.pl.violin(cl1, genes1, groupby='sample')
sc.pl.violin(cl1, genes2, groupby='sample')
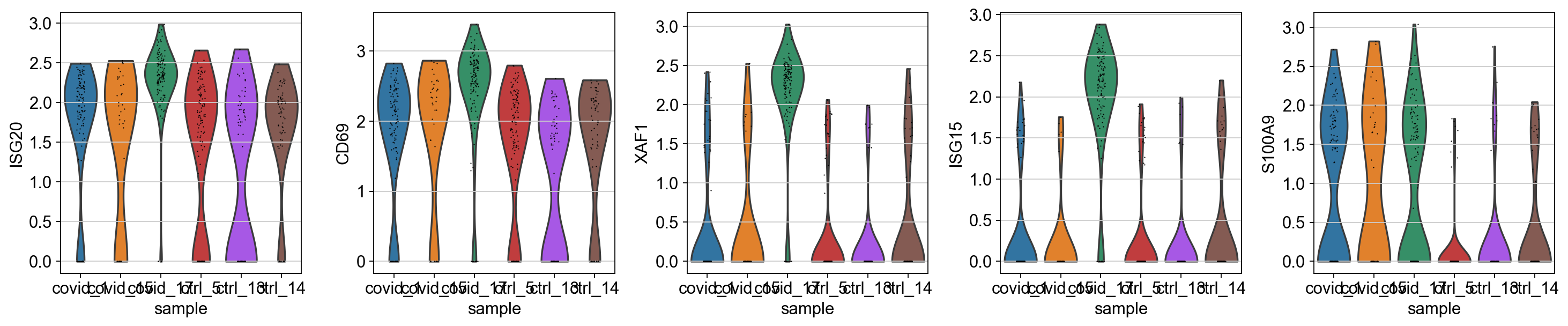
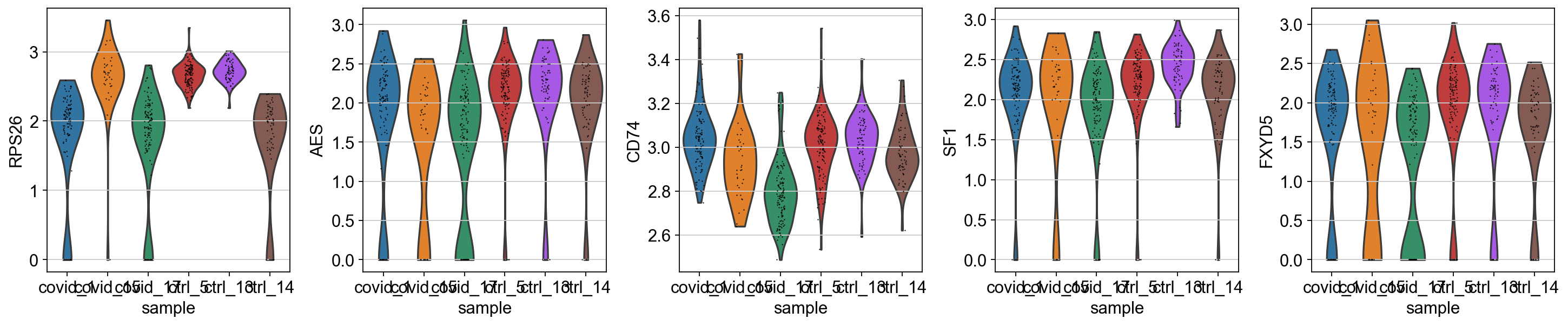
As you can see, many of the genes detected as DGE in Covid are unique to one or 2 patients.
We can examine more genes with a DotPlot:
We can also plot the top Covid and top Ctrl genes as a dotplot:
genes1 = sc.get.rank_genes_groups_df(cl1, group='Covid', key='wilcoxon')['names'][:20]
genes2 = sc.get.rank_genes_groups_df(cl1, group='Ctrl', key='wilcoxon')['names'][:20]
genes = genes1.tolist() + genes2.tolist()
sc.pl.dotplot(cl1,genes, groupby='sample')
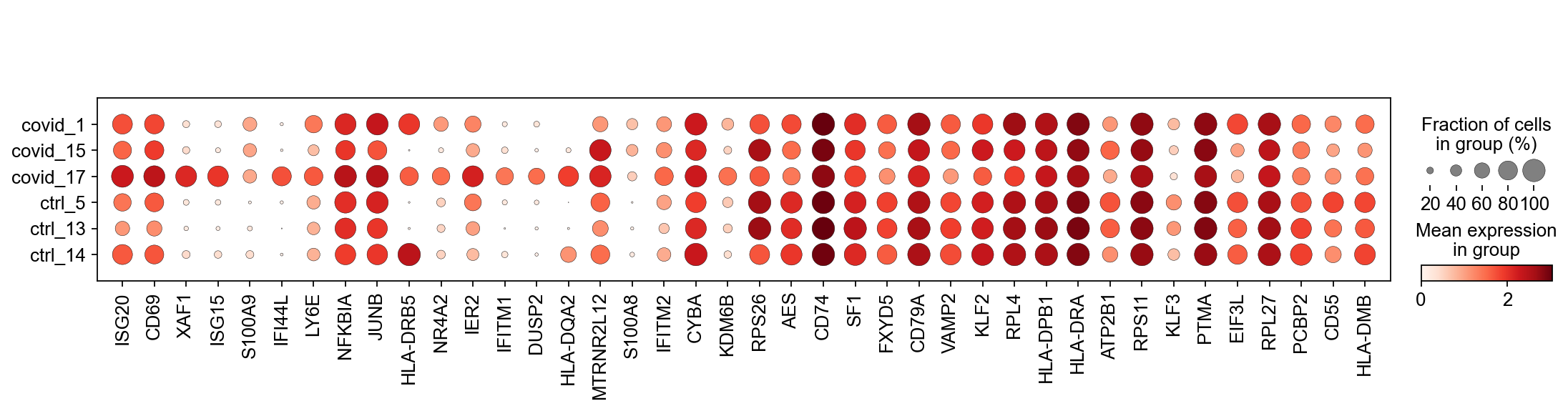
Clearly many of the top Covid genes are only high in the covid_17 sample, and not a general feature of covid patients.
This is also the patient with the highest number of cells in this cluster:
cl1.obs['sample'].value_counts()
Subsample¶
So one obvious thing to consider is an equal amount of cells per individual so that the DGE results are not dominated by a single sample.
So we will downsample to an equal number of cells per sample.
cl1.obs['sample'].value_counts()
target_cells = 50
tmp = [cl1[cl1.obs['sample'] == s] for s in cl1.obs['sample'].cat.categories]
for dat in tmp:
if dat.n_obs > target_cells:
sc.pp.subsample(dat, n_obs=target_cells)
cl1_sub = tmp[0].concatenate(*tmp[1:])
cl1_sub.obs['sample'].value_counts()
sc.tl.rank_genes_groups(cl1_sub, 'type', method='wilcoxon', key_added = "wilcoxon")
sc.pl.rank_genes_groups(cl1_sub, n_genes=25, sharey=False, key="wilcoxon")
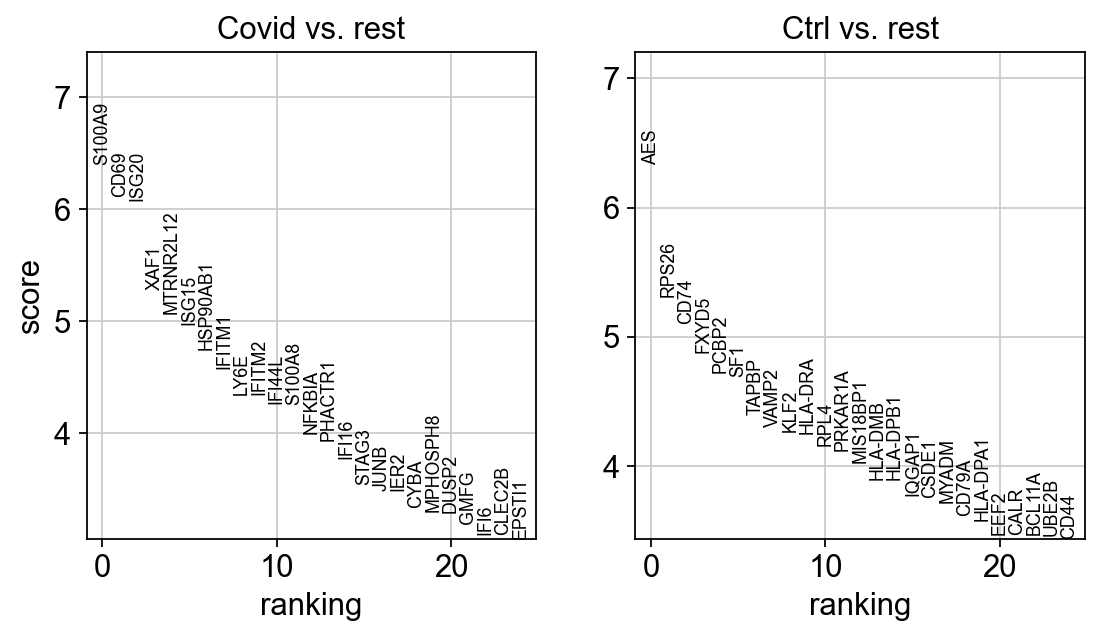
genes1 = sc.get.rank_genes_groups_df(cl1_sub, group='Covid', key='wilcoxon')['names'][:20]
genes2 = sc.get.rank_genes_groups_df(cl1_sub, group='Ctrl', key='wilcoxon')['names'][:20]
genes = genes1.tolist() + genes2.tolist()
sc.pl.dotplot(cl1,genes, groupby='sample')
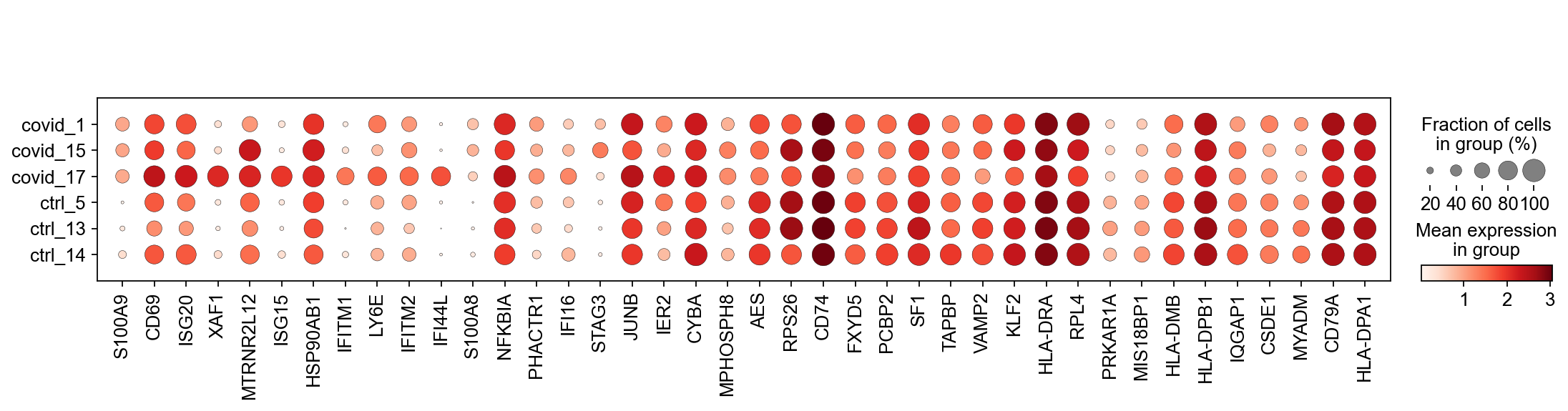
It looks much better now. But if we look per patient you can see that we still have some genes that are dominated by a single patient. Still, it is often a good idea to control the number of cells from each sample when doing differential expression.
Why do you think this is?
There are many different ways to try and resolve the issue of patient batch effects, however most of them require R packages. These can be run via rpy2 as is demonstraded in this compendium: https://www.sc-best-practices.org/conditions/differential_gene_expression.html#
However, we have not included it here as of now. So please have a look at the patient batch effect section in the seurat DGE tutorial where we run EdgeR on pseudobulk and MAST with random effect.
Gene Set Analysis¶
Hypergeometric enrichment test
Having a defined list of differentially expressed genes, you can now look for their combined function using hypergeometric test:
#Available databases : ‘Human’, ‘Mouse’, ‘Yeast’, ‘Fly’, ‘Fish’, ‘Worm’
gene_set_names = gseapy.get_library_name(organism='Human')
print(gene_set_names)
Get the significant DEGs for the Covid patients.
#?gseapy.enrichr
glist = sc.get.rank_genes_groups_df(cl1_sub, group='Covid',
key='wilcoxon', log2fc_min=0.25,
pval_cutoff=0.05)['names'].squeeze().str.strip().tolist()
print(len(glist))
enr_res = gseapy.enrichr(gene_list=glist,
organism='Human',
gene_sets='GO_Biological_Process_2018',
cutoff = 0.5)
enr_res.results.head()
Some databases of interest:
GO_Biological_Process_2017bKEGG_2019_HumanKEGG_2019_MouseWikiPathways_2019_HumanWikiPathways_2019_Mouse
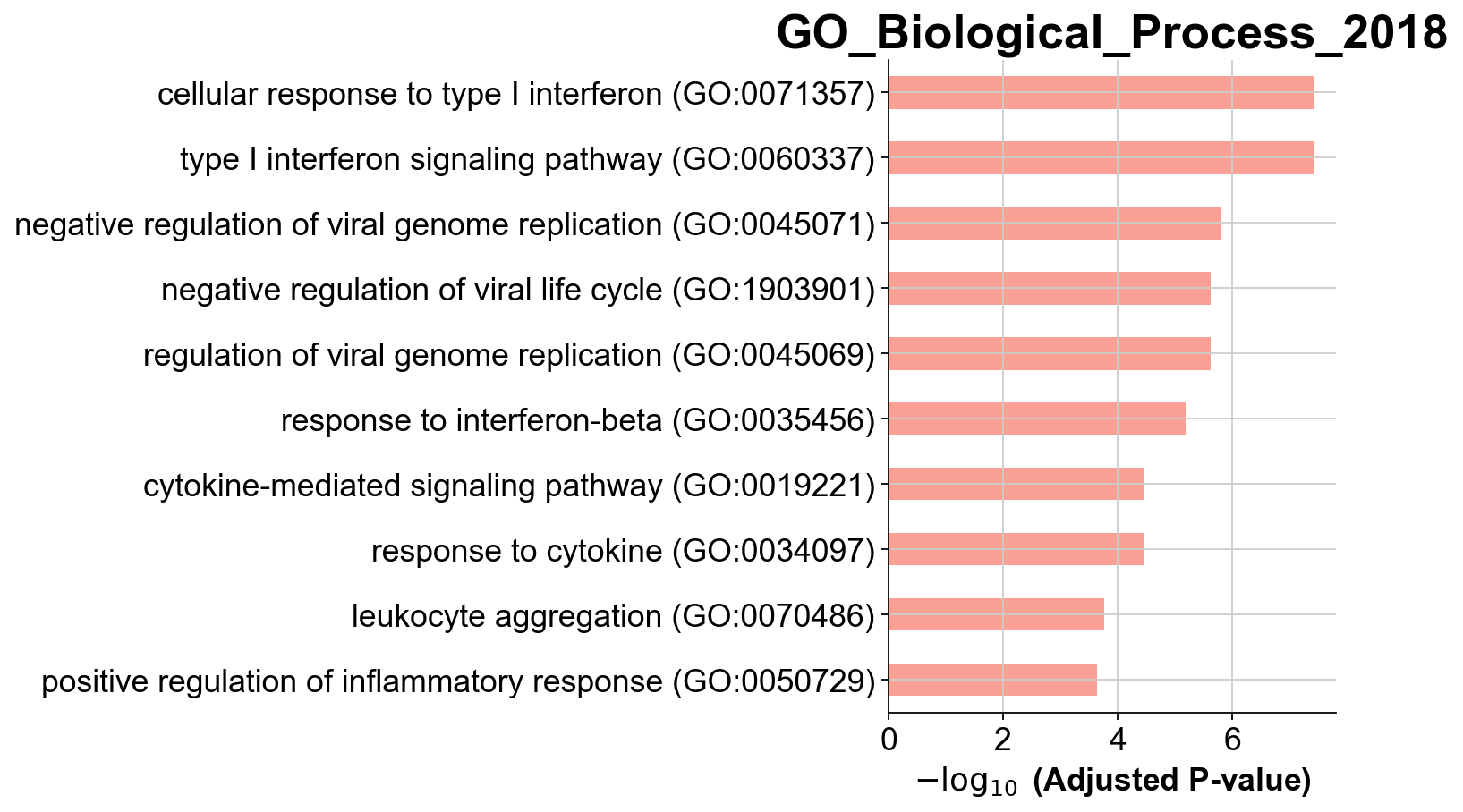
You visualize your results using a simple barplot, for example:
gseapy.barplot(enr_res.res2d,title='GO_Biological_Process_2018')
Gene Set Enrichment Analysis (GSEA)¶
Besides the enrichment using hypergeometric test, we can also perform gene set enrichment analysis (GSEA), which scores ranked genes list (usually based on fold changes) and computes permutation test to check if a particular gene set is more present in the Up-regulated genes, amongthe DOWN_regulated genes or not differentially regulated.
We need a table with all DEGs and their log foldchanges. However, many lowly expressed genes will have high foldchanges and just contribue noise, so also filter for expression in enough cells.
gene_rank = sc.get.rank_genes_groups_df(cl1_sub, group='Covid', key='wilcoxon')[['names','logfoldchanges']]
gene_rank.sort_values(by=['logfoldchanges'], inplace=True, ascending=False)
# calculate_qc_metrics will calculate number of cells per gene
sc.pp.calculate_qc_metrics(cl1, percent_top=None, log1p=False, inplace=True)
# filter for genes expressed in at least 30 cells.
gene_rank = gene_rank[gene_rank['names'].isin(cl1.var_names[cl1.var.n_cells_by_counts>30])]
gene_rank
Once our list of genes are sorted, we can proceed with the enrichment itself. We can use the package to get gene set from the Molecular Signature Database (MSigDB) and select KEGG pathways as an example.
#Available databases : ‘Human’, ‘Mouse’, ‘Yeast’, ‘Fly’, ‘Fish’, ‘Worm’
gene_set_names = gseapy.get_library_name(organism='Human')
print(gene_set_names)
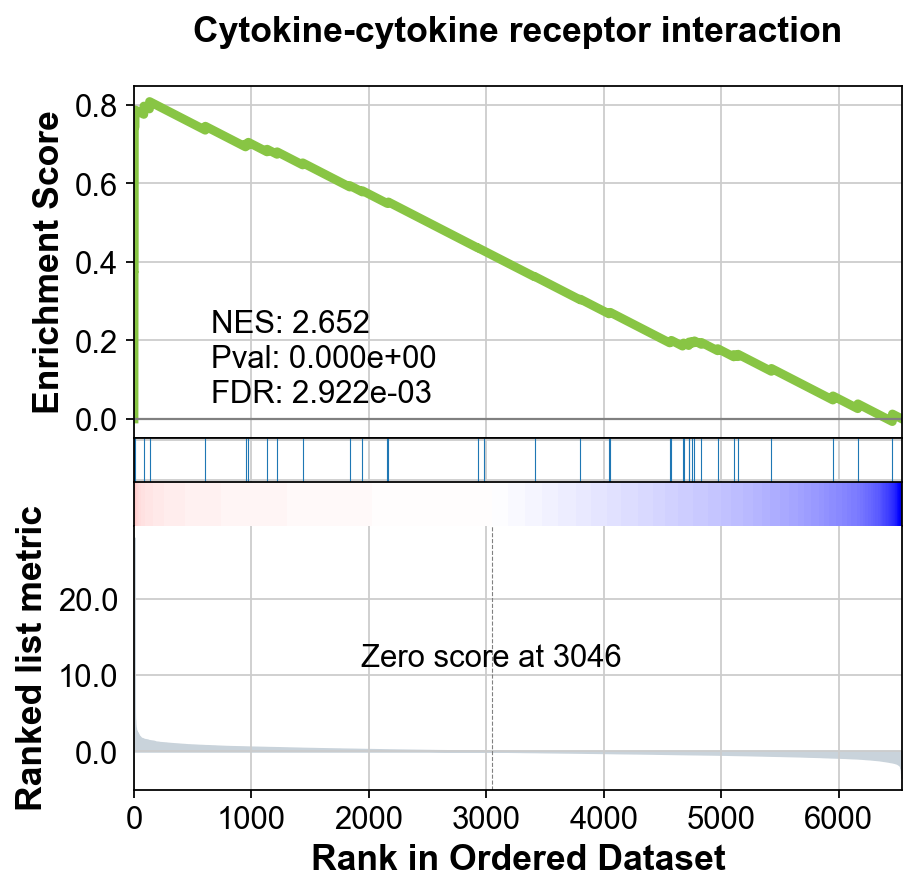
Next, we will be using the GSEA. This will result in a table containing information for several pathways. We can then sort and filter those pathways to visualize only the top ones. You can select/filter them by either p-value or normalized enrichemnet score (NES).
res = gseapy.prerank(rnk=gene_rank, gene_sets='KEGG_2021_Human')
terms = res.res2d.Term
terms[:10]
gseapy.gseaplot(rank_metric=res.ranking, term=terms[0], **res.results[terms[0]])
Finally, lets save the integrated data for further analysis.
adata.write_h5ad('./data/results/scanpy_DGE_covid.h5ad')