class: center, middle, inverse, title-slide # Differential Gene Expression Workflow ## Workshop on RNA-Seq ### <b>Roy Francis</b> | 03-Jun-2019 --- exclude: true count: false <link href="https://fonts.googleapis.com/css?family=Roboto|Source+Sans+Pro:300,400,600|Ubuntu+Mono&subset=latin-ext" rel="stylesheet"> <link rel="stylesheet" href="https://use.fontawesome.com/releases/v5.3.1/css/all.css" integrity="sha384-mzrmE5qonljUremFsqc01SB46JvROS7bZs3IO2EmfFsd15uHvIt+Y8vEf7N7fWAU" crossorigin="anonymous"> <!-- ------------ Only edit title, subtitle & author above this ------------ --> --- name: content class: spaced ## Contents * [Preprocessing](#pp) * [Normalisation](#norm-1) * [Exploratory](#eda-cor) * [DGE](#dge) --- name: raw ## Raw data - Raw count table ``` ## Sample_1 Sample_2 Sample_3 Sample_4 Sample_5 Sample_6 ## ENSG00000000003 321 303 204 492 455 359 ## ENSG00000000005 0 0 0 0 0 0 ## ENSG00000000419 696 660 472 951 963 689 ## ENSG00000000457 59 54 44 109 73 66 ## ENSG00000000460 399 405 236 445 454 374 ## ENSG00000000938 0 0 0 0 0 1 ``` - Metadata ``` ## Sample_ID Sample_Name Time Replicate Cell ## Sample_1 Sample_1 t0_A t0 A A431 ## Sample_2 Sample_2 t0_B t0 B A431 ## Sample_3 Sample_3 t0_C t0 C A431 ## Sample_4 Sample_4 t2_A t2 A A431 ## Sample_5 Sample_5 t2_B t2 B A431 ## Sample_6 Sample_6 t2_C t2 C A431 ``` --- name: pp ## Preprocessing - Remove genes and samples with low counts ```r cf1 <- cr[rowSums(cr>0) >= 2, ] cf2 <- cr[rowSums(cr>5) >= 2, ] cf3 <- cr[rowSums(edgeR::cpm(cr)>1) >= 2, ] ``` -- - Inspect distribution <img src="presentation_dge_files/figure-html/unnamed-chunk-7-1.svg" style="display: block; margin: auto auto auto 0;" /> -- - Inspect the number of rows ``` ## Raw: 59573, Method 1: 24194, Method 2: 16519, Method 3: 14578 ``` --- name: norm-1 ## Normalisation .pull-left-50[ - Make counts comparable across samples - Control for sequencing depth 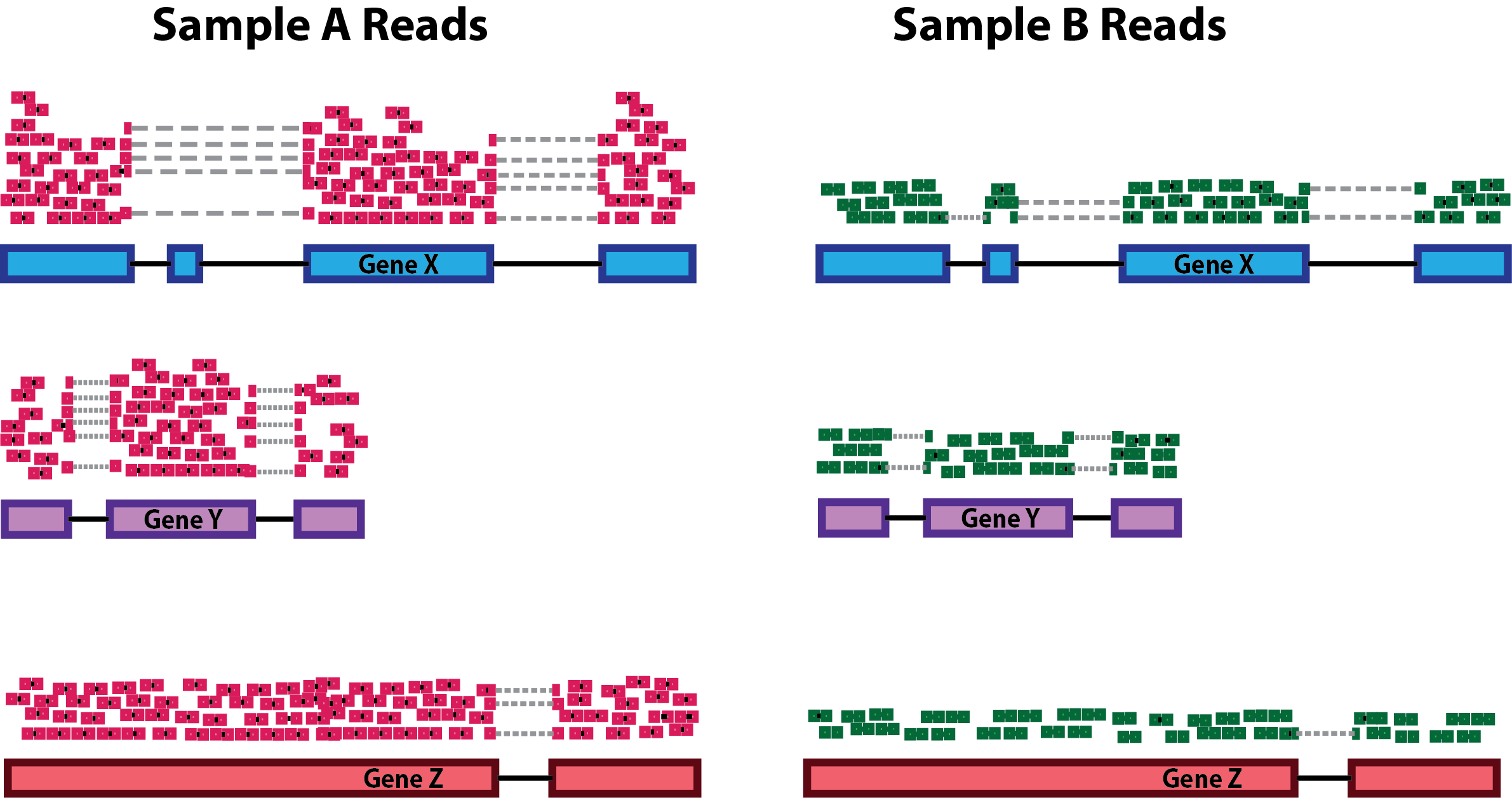 ``` ## s1 s2 s1_tc s2_tc ## x 20 6 0.33 0.38 ## y 25 6 0.42 0.38 ## z 15 4 0.25 0.25 ``` ] -- .pull-right-50[ - Control for compositional bias  ``` ## s1 s2 s1_tc s2_tc ## x 20 20 0.12 0.39 ## y 25 25 0.16 0.49 ## z 15 4 0.09 0.08 ## de 100 2 0.62 0.04 ``` ] --- name: norm-2 ## Normalisation - Make counts comparable across features (genes) .size-60[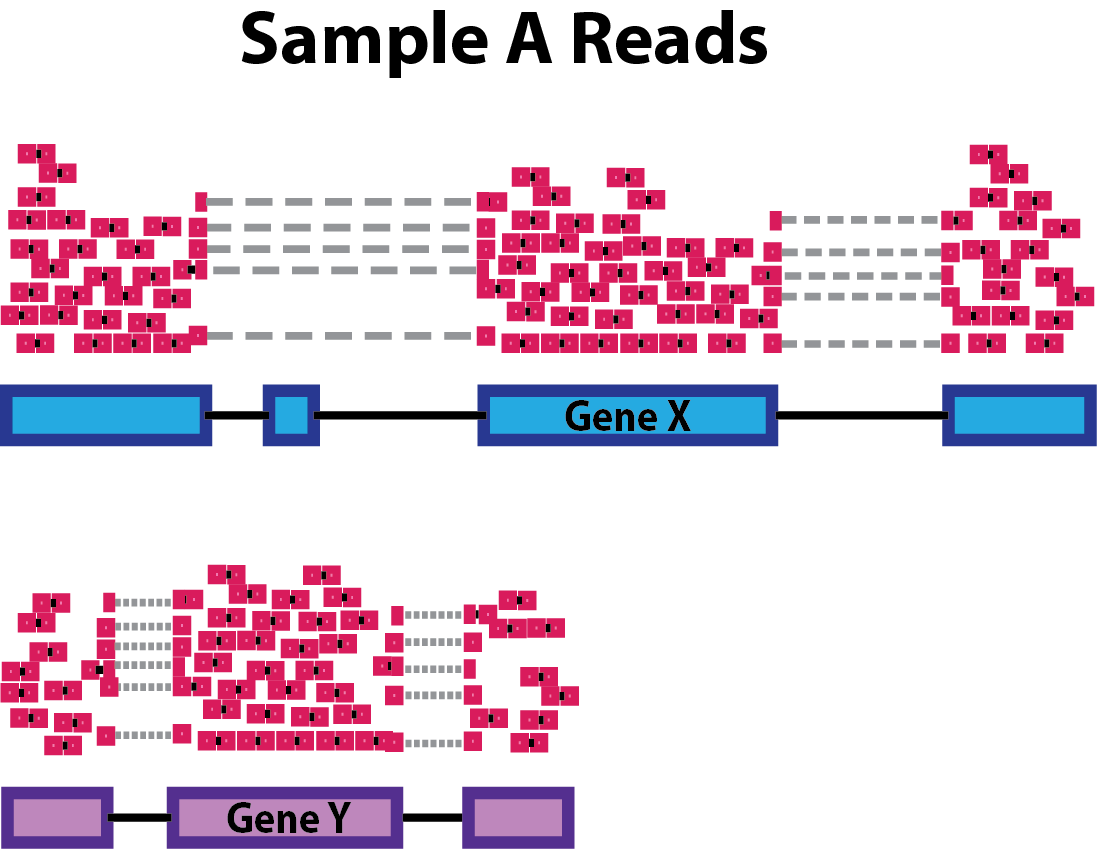] ``` ## counts gene_length norm_counts ## 1 50 10 5 ## 2 25 5 5 ``` -- - Bring counts to a human-friendly scale --- name: norm-3 ## Normalisation - CPM, RPKM, FPKM, TPM, RLE, MRN, Q, UQ, TMM, VST, RLOG, VOOM ... Too many... -- - **CPM**: Controls for sequencing depth when dividing by total count. Not for within-sample comparison or DE. -- - **RPKM/FPKM**: Controls for sequencing depth and gene length. Good for technical replicates, not good for sample-sample due to compositional bias. Assumes total RNA output is same in all samples. Not for DE. -- - **TPM**: Similar to RPKM/FPKM. Corrects for sequencing depth and gene length. Also comparable between samples but no correction for compositional bias. -- - **TMM/RLE/MRN**: Improved assumption: The output between samples for a core set only of genes is similar. Corrects for compositional bias. Used for DE. RLE and MRN are very similar and correlates well with sequencing depth. `edgeR::calcNormFactors()` implements TMM, TMMwzp, RLE & UQ. `DESeq2::estimateSizeFactors` implements median ratio method (RLE). Does not correct for gene length. -- - **VST/RLOG/VOOM**: Variance is stabilised across the range of mean values. For use in exploratory analyses. Not for DE. `vst()` and `rlog()` functions from *DESeq2*. `voom()` function from *Limma* converts data to normal distribution. -- - **[geTMM](https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2246-7)**: Gene length corrected TMM. .citation[ .cite[<i class="fas fa-link"></i> Dillies, Marie-Agnes, *et al*. "A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis." [Briefings in bioinformatics 14.6 (2013): 671-683](https://www.ncbi.nlm.nih.gov/pubmed/22988256)] .cite[<i class="fas fa-link"></i> Evans, Ciaran, Johanna Hardin, and Daniel M. Stoebel. "Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions." [Briefings in bioinformatics (2017)](https://arxiv.org/abs/1609.00959)] ] --- name: norm-4 ## Normalisation **Recommendations** - For DGE using DGE R packages (DESeq2, edgeR, Limma etc), use raw counts - For visualisation (PCA, clustering, heatmaps etc), use VST or RLOG - For own analysis with gene length correction, use TPM (maybe geTMM?) - Other solutions: spike-ins/house-keeping genes -- <img src="presentation_dge_files/figure-html/unnamed-chunk-13-1.svg" style="display: block; margin: auto auto auto 0;" /> <img src="presentation_dge_files/figure-html/unnamed-chunk-14-1.svg" style="display: block; margin: auto auto auto 0;" /> --- name: eda-cor ## Exploratory | Correlation * Correlation between samples ```r dmat <- as.matrix(cor(cv,method="spearman")) pheatmap::pheatmap(dmat,border_color=NA,annotation_col=mr[,"Time",drop=F], annotation_row=mr[,"Time",drop=F],annotation_legend=T) ``` --- name: eda-dist ## Exploratory | Distance * Similarity between samples ```r dmat <- as.matrix(dist(t(cv))) pheatmap(dmat,border_color=NA,annotation_col=mr[,"Time",drop=F], annotation_row=mr[,"Time",drop=F],annotation_legend=T) ``` --- name: eda-pca ## Exploratory | PCA * Relationship between samples .pull-left-40[ <img src="presentation_dge_files/figure-html/unnamed-chunk-17-1.svg" style="display: block; margin: auto auto auto 0;" /> ] .pull-right-60[ <img src="presentation_dge_files/figure-html/unnamed-chunk-18-1.svg" style="display: block; margin: auto auto auto 0;" /> ] --- name: dge ## DGE * Create the DESeq2 object ```r library(DESeq2) mr$Time <- factor(mr$Time) d <- DESeqDataSetFromMatrix(countData=cf,colData=mr,design=~Time) d ``` ``` ## class: DESeqDataSet ## dim: 14578 12 ## metadata(1): version ## assays(1): counts ## rownames(14578): ENSG00000000003 ENSG00000000419 ... ## ENSG00000266865 ENSG00000266876 ## rowData names(0): ## colnames(12): Sample_1 Sample_2 ... Sample_11 Sample_12 ## colData names(5): Sample_ID Sample_Name Time Replicate Cell ``` * Model must be factors - `~var` - `~covar+var` --- name: dge-sf ## DGE | Size factors * Normalisation factors are computed ```r d <- DESeq2::estimateSizeFactors(d,type="ratio") sizeFactors(d) ``` ``` ## Sample_1 Sample_2 Sample_3 Sample_4 Sample_5 Sample_6 Sample_7 ## 0.9003753 0.8437393 0.5106445 1.1276451 1.0941383 0.8133849 0.7553903 ## Sample_8 Sample_9 Sample_10 Sample_11 Sample_12 ## 1.1744008 1.0189325 1.3642797 1.2325485 1.8555904 ``` --- name: dge-dispersion ## DGE | Dispersion * We need a measure variability of gene counts ```r dm <- apply(cd,1,mean) dv <- apply(cd,1,var) cva <- function(x) sd(x)/mean(x) dc <- apply(cd,1,cva) ``` -- <img src="presentation_dge_files/figure-html/unnamed-chunk-23-1.svg" style="display: block; margin: auto auto auto 0;" /> * Dispersion is a measure of variability in gene expression for a given mean --- name: dge-dispersion-2 ## DGE | Dispersion * D is unreliable for low mean counts * Genes with similar mean values must have similar dispersion * Estimate likely (ML) dispersion for each gene based on counts * Fit a curve through the gene-wise estimates * Shrink dispersion towards the curve ```r d <- DESeq2::estimateDispersions(d) {par(mar=c(4,4,1,1)) plotDispEsts(d)} ``` <img src="presentation_dge_files/figure-html/unnamed-chunk-24-1.svg" style="display: block; margin: auto auto auto 0;" /> --- name: dge-test ## DGE | Test * Log2 fold changes changes are computed after GLM fitting ```r dg <- nbinomWaldTest(d) resultsNames(dg) ``` ``` ## [1] "Intercept" "Time_t2_vs_t0" "Time_t24_vs_t0" "Time_t6_vs_t0" ``` -- * Use `results()` to customise/return results * Set coefficients using `contrast` or `name` * Filtering by fold change using `lfcThreshold` * `cooksCutoff` removes outliers * `independentFiltering` * `pAdjustMethod` * `alpha` --- name: dge-test-2 ## DGE | Test ```r res1 <- results(dg,name="Time_t2_vs_t0",alpha=0.05) summary(res1) ``` ``` ## ## out of 14578 with nonzero total read count ## adjusted p-value < 0.05 ## LFC > 0 (up) : 413, 2.8% ## LFC < 0 (down) : 696, 4.8% ## outliers [1] : 0, 0% ## low counts [2] : 2261, 16% ## (mean count < 26) ## [1] see 'cooksCutoff' argument of ?results ## [2] see 'independentFiltering' argument of ?results ``` --- name: dge-test-3 ## DGE | Test ```r head(res1) ``` ``` ## log2 fold change (MLE): Time t2 vs t0 ## Wald test p-value: Time t2 vs t0 ## DataFrame with 6 rows and 6 columns ## baseMean log2FoldChange lfcSE ## <numeric> <numeric> <numeric> ## ENSG00000000003 490.017213130775 0.220619809383656 0.112761083788383 ## ENSG00000000419 817.780657499214 0.0592719659601406 0.101481284662652 ## ENSG00000000457 82.078767893562 0.207748623172909 0.220404896514001 ## ENSG00000000460 356.07160020304 -0.129186381676072 0.115139182335103 ## ENSG00000001036 919.606750211436 0.0288826917736355 0.0851500691928604 ## ENSG00000001084 529.593965301543 0.211964772147193 0.0929810563117334 ## stat pvalue padj ## <numeric> <numeric> <numeric> ## ENSG00000000003 1.95652437854969 0.0504034135093116 0.263505451695327 ## ENSG00000000419 0.584067950629267 0.559174596319379 0.830262316260423 ## ENSG00000000457 0.942577168015466 0.34589722283592 0.689945926100409 ## ENSG00000000460 -1.12200190288033 0.2618616308297 0.612624613846088 ## ENSG00000001036 0.33919751384133 0.734460942036292 0.909638554355053 ## ENSG00000001084 2.27965545408033 0.0226281312376226 0.159263252830742 ``` -- * Use `lfcShrink()` to correct fold changes for high dispersion genes <!-- --------------------- Do not edit this and below --------------------- --> --- # Acknowledgements * [Normalising RNA-seq data](https://www.ebi.ac.uk/sites/ebi.ac.uk/files/content.ebi.ac.uk/materials/2012/121029_HTS/ernest_turro_normalising_rna-seq_data.pdf) by Ernest Turro * RNA-seq analysis [Bioconductor vignette](http://master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html) * [DGE Workshop](https://github.com/hbctraining/DGE_workshop/tree/master/lessons) by HBC training --- name: end_slide class: end-slide, middle count: false # Thank you. Questions? .end-text[ <p>R version 3.5.2 (2018-12-20)<br><p>Platform: x86_64-pc-linux-gnu (64-bit)</p><p>OS: Ubuntu 18.04.2 LTS</p><br> <hr> <span class="small">Built on : <i class='fa fa-calendar' aria-hidden='true'></i> 03-Jun-2019 at <i class='fa fa-clock-o' aria-hidden='true'></i> 22:54:57</span> <b>2019</b> • [SciLifeLab](https://www.scilifelab.se/) • [NBIS](https://nbis.se/) ]