Cell type identification
Created by: Philip Lijnzaad
Overview
In any single-cell experiment where mixed populations are expected, a very important task is to identify the cell types (and/or cell states) that are present. In this practical, we will look at two tools for doing this: SingleR (Aran et al. 2019) and CHETAH (de Kanter et al., under review).
Datasets
We will try to classify the ovarian ascites data from Schelker et al. 2017 using, as a reference, the different reference data sets from both SingleR and CHETAH.
The ovarian1200 dataset contains 1200 cells randomly selected from the
3114 single cell data provided by Schelker et al. (which is available
from https://figshare.com/s/711d3fb2bd3288c8483). The data come from
the ascites of 4 different ovarian cancer patients and contain a variety
of leukocytes as well as tumor cells.
First load the various packages:
suppressMessages(require(SingleCellExperiment))
suppressMessages(require(Seurat))
suppressMessages(require(CHETAH))
suppressMessages(require(Matrix))
suppressMessages(require(SingleR))
suppressMessages(require(cowplot))
If you have (or downloaded) the ovarian data into folder data.dir then
load the Seurat object with
#load expression matrix
data.dir <- "session-celltypeid_files" # or wherever the data is located
file <- paste0(data.dir,"/ovarian1200.rds")
ovarian <- readRDS(file=file)
The authors already classified the cells ‘by hand’ using marker genes.
This classfication can be found in the celltypes column of the
meta.data of the object. Get an overview of this:
head(ovarian@meta.data)
## nGene nUMI orig.ident celltypes nCount_RNA nFeature_RNA
## X7873M.1 3563 11796 SeuratProject Macrophage 11796 3563
## X7873M.104 2670 7570 SeuratProject Macrophage 7570 2670
## X7873M.105 2241 5510 SeuratProject Macrophage 5510 2241
## X7873M.106 4705 22636 SeuratProject CAF 22636 4705
## X7873M.110 3169 11690 SeuratProject Macrophage 11690 3169
## X7873M.114 4659 20798 SeuratProject Macrophage 20798 4659
sort(table(ovarian@meta.data$celltypes))
##
## reg. T cell B cell CAF CD8 T cell NK Dendritic
## 0 7 9 10 28 46
## Unknown Tumor CD4 T cell Macrophage
## 50 117 129 804
Clearly, most of the cells are Macrophages.
We now follow the ‘standard’ Seurat workflow to prepare the data.
ovarian <- NormalizeData(object = ovarian)
ovarian <- FindVariableFeatures(object = ovarian)
ovarian <- ScaleData(object = ovarian)
## Centering and scaling data matrix
ovarian <- RunPCA(object = ovarian, npcs=20)
## PC_ 1
## Positive: KRT19, TACSTD2, SLPI, KRT18, LCN2, KRT7, CRYAB, TM4SF1, MAL2, S100A16
## PXDN, WFDC2, MUC4, CLDN1, C19orf33, KRT18P55, GNG12, CLDN4, UCA1, PERP
## MDK, KLK6, CNN3, CLDN3, KIAA1522, DCBLD2, TSPAN1, NBL1, CTTN, SCNN1A
## Negative: FTL, HLA-DRA, LYZ, VCAN, S100A8, VSIG4, FABP5, HLA-DRB1, SPP1, HLA-DPA1
## MARCO, HLA-DPB1, CTSL, HLA-DQB1, CD36, CCL2, CTSB, TIMP1, C1QA, S100A9
## EPB41L3, FGR, LGMN, RNASE1, CD300E, SELL, CLEC5A, IFI6, HLA-DQA1, OLR1
## PC_ 2
## Positive: EGFL6, C1S, C1R, CEMIP, CALB2, SPARC, ANXA8, COL3A1, C4B, C4A
## ANXA8L1, COL1A2, PTGIS, COL6A1, XXbac-BPG116M5.17, MXRA8, IGFBP4, GJA1, CFB, GRIA2
## MGP, SOD3, MEDAG, COL5A2, CALD1, RARRES2, CCDC80, ALDH1A2, CD248, PLA2G2A
## Negative: TACSTD2, LCN2, MUC4, UCA1, MAL2, CAMK2N1, LAD1, CLDN4, SPINT1, VSTM2L
## MAL, CLDN3, SCNN1A, KLK6, TSPAN1, SPOCK2, CCND1, ELF3, EPCAM, RAB25
## MUC20, CD24, NTN4, KRT80, PRSS8, KLK7, SMIM22, BHLHE41, PERP, KRT23
## PC_ 3
## Positive: CTSB, CTSL, FTL, VSIG4, FABP5, MARCO, SPP1, CTSD, TIMP1, PLTP
## FN1, S100A9, RNASE1, CCL2, VCAN, FTH1, C1QA, ANXA2, S100A8, C1QB
## C1QC, LGMN, HLA-DRB1, LYVE1, IFI6, HLA-DRA, DAB2, GPNMB, LYZ, EPB41L3
## Negative: LTB, TRBC2, IL32, IFITM1, TRBC1, MIAT, IL7R, SLC38A1, TRAC, CCL5
## BCL11B, GZMK, SPOCK2, GZMB, CXCR4, IL2RB, CD2, JCHAIN, FYN, SELL
## DERL3, GZMA, LILRA4, CCR7, ITM2C, C12orf75, IGKC, CST7, KLRB1, PIM2
## PC_ 4
## Positive: TUBB2B, UCHL1, MYBL2, UBE2C, CTHRC1, CRABP2, PEG10, TPX2, CKS1B, CDKN2A
## AURKB, CKB, KIFC1, PRC1, TYMS, TK1, CDCA8, TOP2A, IGFBP2, MEX3A
## STMN1, NOV, PTTG1, CDK1, MKI67, SMPDL3B, CENPM, CDC20, ANLN, PLAU
## Negative: VSTM2L, C10orf10, PDZK1IP1, CAMK2N1, LMO7, MUC4, MSLN, MUC21, VWA5A, GPRC5A
## LCN2, MAL, FXYD3, MESP1, DHRS9, SPOCK2, SAT1, C3, AHNAK, TGM2
## CD55, KLK11, HOPX, TTC9, MUC20, C1orf116, SNCG, EVPL, GPC1, FAM3C
## PC_ 5
## Positive: DERL3, GZMB, LILRA4, JCHAIN, SERPINF1, PLD4, IGKC, IRF8, TSPAN13, IRF7
## TCF4, SCAMP5, CYB561A3, LRRC26, ITM2C, SMPD3, LAMP5, HLA-DPA1, HLA-DRA, CCDC50
## IGHM, GPM6B, TCL1A, PLAC8, HLA-DPB1, IL3RA, HLA-DQA1, HLA-DRB1, C12orf75, HLA-DQB1
## Negative: MTRNR2L12, MTRNR2L8, IL32, TRBC2, MIAT, IL7R, TRBC1, BCL11B, MTRNR2L1, CCL5
## TRAC, MT-ND4L, IFITM1, GZMK, IL2RB, MTRNR2L6, SPOCK2, CD2, MTRNR2L3, GZMA
## NEAT1, KLRB1, CDKN2A, ITM2A, FYN, CD8A, MTRNR2L11, SIX3, ARL4C, UCHL1
ovarian <- FindNeighbors(object = ovarian)
## Computing nearest neighbor graph
## Computing SNN
ovarian <- FindClusters(object = ovarian, resolution=0.5)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 1200
## Number of edges: 37767
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8624
## Number of communities: 9
## Elapsed time: 0 seconds
## (the default resolution yields too manh clusters)
ovarian <- RunTSNE(object = ovarian)
ovarian <- RunUMAP(object = ovarian, dims=1:20)
p1 <- DimPlot(ovarian, reduction='tsne')
p2 <- DimPlot(ovarian, reduction='umap')
plot_grid(p1, p2)
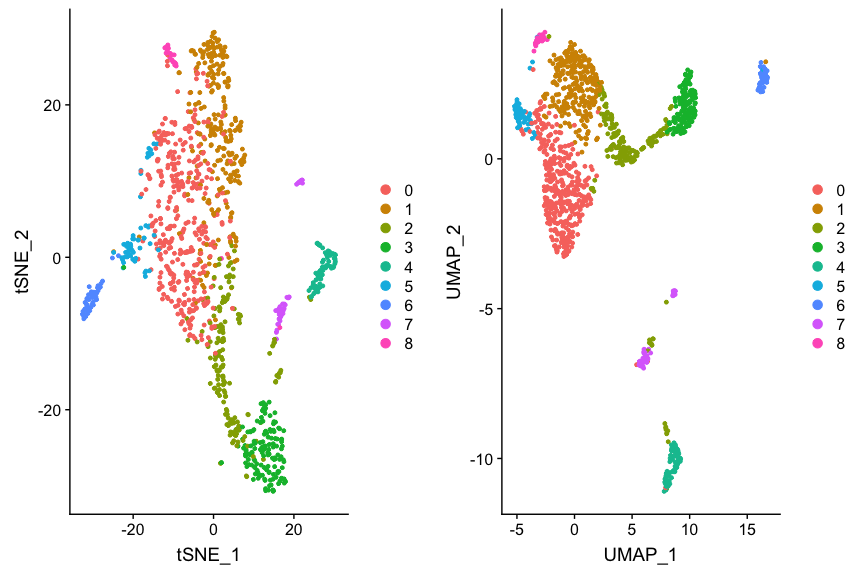
This shows the data with Seurat clustering, but we’re more interested in
the cell types. Let’s see if and how they coincide. For convenience and
flexibility, we define dim.red to be our dimension reduction of
choice.
dim.red <- 'tsne' # or 'umap' or 'pca'
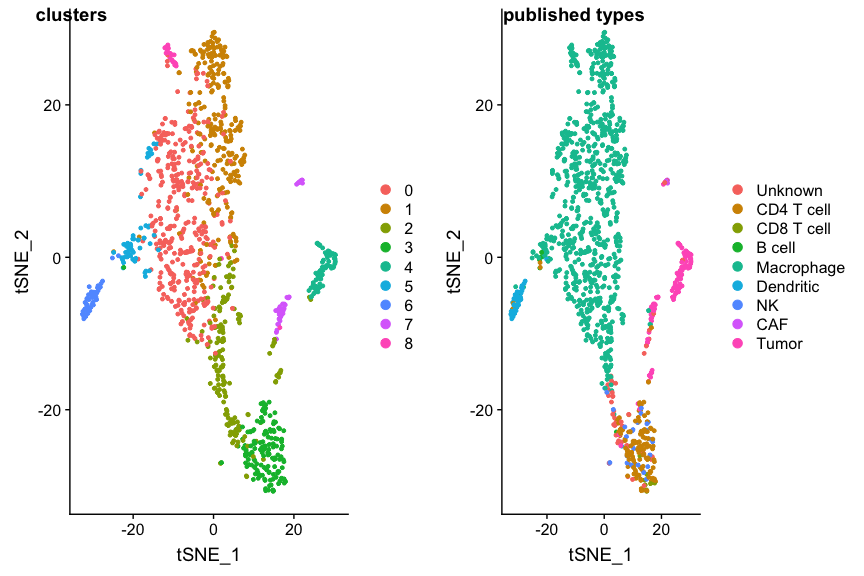
p1 <- DimPlot(ovarian, group.by="seurat_clusters", reduction=dim.red)
p2 <- DimPlot(ovarian, group.by='celltypes', reduction=dim.red)
plot_grid(p1, p2, labels=c('clusters', 'published types'))
SingleR
SingleR reference data sets
We will now do our cell type identification using SingleR. SingleR comes
with a collection of reference data sets. There are two for human:
hpca, the Human Primary Cell Atlas (microarray-based), and
blueprint\_encode, a combined Blueprint Epigenomics and Encode data
set (RNASeq based) .
For mouse there are also two sets: immgen, the Immunological Genome
Project (microarray-based) and mouse.rnaseq, a brain specific
reference (RNASeq-based). For details I refer to the
SingleR-specifications vignette.
Each reference data set comes in two ‘flavours’: ‘types’, which are very fine-grained, detailed types, and ‘main_types’, which are less coarser subset of those types.
The SingleR reference data sets are part of the package, and can be explored easily.
table(hpca$main_types)
##
## Astrocyte BM BM & Prog.
## 2 7 1
## B_cell CMP Chondrocytes
## 26 2 8
## DC Embryonic_stem_cells Endothelial_cells
## 88 17 64
## Epithelial_cells Erythroblast Fibroblasts
## 16 8 10
## GMP Gametocytes HSC_-G-CSF
## 2 5 10
## HSC_CD34+ Hepatocytes Keratinocytes
## 6 3 25
## MEP MSC Macrophage
## 2 9 90
## Monocyte Myelocyte NK_cell
## 60 2 5
## Neuroepithelial_cell Neurons Neutrophil
## 1 16 3
## Neutrophils Osteoblasts Platelets
## 18 15 5
## Pre-B_cell_CD34- Pro-B_cell_CD34+ Pro-Myelocyte
## 2 2 2
## Smooth_muscle_cells T_cells Tissue_stem_cells
## 16 68 55
## iPS_cells
## 42
table(hpca$types)
##
## Astrocyte:Embryonic_stem_cell-derived
## 2
## BM
## 8
## B_cell
## 4
## B_cell:CXCR4+_centroblast
## 4
## B_cell:CXCR4-_centrocyte
## 4
## B_cell:Germinal_center
## 3
## B_cell:Memory
## 3
## B_cell:Naive
## 3
## B_cell:Plasma_cell
## 3
## B_cell:immature
## 2
## CMP
## 2
## Chondrocytes:MSC-derived
## 8
## DC:monocyte-derived
## 19
## DC:monocyte-derived:A._fumigatus_germ_tubes_6h
## 2
## DC:monocyte-derived:AEC-conditioned
## 5
## DC:monocyte-derived:AM580
## 3
## DC:monocyte-derived:CD40L
## 3
## DC:monocyte-derived:Galectin-1
## 3
## DC:monocyte-derived:LPS
## 6
## DC:monocyte-derived:Poly(IC)
## 3
## DC:monocyte-derived:Schuler_treatment
## 3
## DC:monocyte-derived:anti-DC-SIGN_2h
## 3
## DC:monocyte-derived:antiCD40/VAF347
## 2
## DC:monocyte-derived:immature
## 20
## DC:monocyte-derived:mature
## 11
## DC:monocyte-derived:rosiglitazone
## 3
## DC:monocyte-derived:rosiglitazone/AGN193109
## 2
## Embryonic_stem_cells
## 17
## Endothelial_cells:HUVEC
## 16
## Endothelial_cells:HUVEC:B._anthracis_LT
## 2
## Endothelial_cells:HUVEC:Borrelia_burgdorferi
## 2
## Endothelial_cells:HUVEC:FPV-infected
## 3
## Endothelial_cells:HUVEC:H5N1-infected
## 3
## Endothelial_cells:HUVEC:IFNg
## 1
## Endothelial_cells:HUVEC:IL-1b
## 3
## Endothelial_cells:HUVEC:PR8-infected
## 3
## Endothelial_cells:HUVEC:Serum_Amyloid_A
## 6
## Endothelial_cells:HUVEC:VEGF
## 3
## Endothelial_cells:blood_vessel
## 8
## Endothelial_cells:lymphatic
## 7
## Endothelial_cells:lymphatic:KSHV
## 4
## Endothelial_cells:lymphatic:TNFa_48h
## 3
## Epithelial_cells:bladder
## 6
## Epithelial_cells:bronchial
## 10
## Erythroblast
## 8
## Fibroblasts:breast
## 6
## Fibroblasts:foreskin
## 4
## GMP
## 2
## Gametocytes:oocyte
## 3
## Gametocytes:spermatocyte
## 2
## HSC_-G-CSF
## 10
## HSC_CD34+
## 6
## Hepatocytes
## 3
## Keratinocytes
## 3
## Keratinocytes:IFNg
## 2
## Keratinocytes:IL19
## 3
## Keratinocytes:IL1b
## 2
## Keratinocytes:IL20
## 3
## Keratinocytes:IL22
## 3
## Keratinocytes:IL24
## 3
## Keratinocytes:IL26
## 3
## Keratinocytes:KGF
## 3
## MEP
## 2
## MSC
## 9
## Macrophage:Alveolar
## 4
## Macrophage:Alveolar:B._anthacis_spores
## 3
## Macrophage:monocyte-derived
## 26
## Macrophage:monocyte-derived:IFNa
## 9
## Macrophage:monocyte-derived:IL-4/Dex/TGFb
## 10
## Macrophage:monocyte-derived:IL-4/Dex/cntrl
## 5
## Macrophage:monocyte-derived:IL-4/TGFb
## 5
## Macrophage:monocyte-derived:IL-4/cntrl
## 5
## Macrophage:monocyte-derived:M-CSF
## 2
## Macrophage:monocyte-derived:M-CSF/IFNg
## 2
## Macrophage:monocyte-derived:M-CSF/IFNg/Pam3Cys
## 2
## Macrophage:monocyte-derived:M-CSF/Pam3Cys
## 2
## Macrophage:monocyte-derived:S._aureus
## 15
## Monocyte
## 27
## Monocyte:CD14+
## 3
## Monocyte:CD16+
## 6
## Monocyte:CD16-
## 7
## Monocyte:CXCL4
## 2
## Monocyte:F._tularensis_novicida
## 6
## Monocyte:MCSF
## 2
## Monocyte:S._typhimurium_flagellin
## 1
## Monocyte:anti-FcgRIIB
## 2
## Monocyte:leukotriene_D4
## 4
## Myelocyte
## 2
## NK_cell
## 1
## NK_cell:CD56hiCD62L+
## 1
## NK_cell:IL2
## 3
## Neuroepithelial_cell:ESC-derived
## 1
## Neurons:ES_cell-derived_neural_precursor
## 6
## Neurons:Schwann_cell
## 4
## Neurons:adrenal_medulla_cell_line
## 6
## Neutrophil
## 6
## Neutrophil:GM-CSF_IFNg
## 4
## Neutrophil:LPS
## 4
## Neutrophil:commensal_E._coli_MG1655
## 2
## Neutrophil:inflam
## 4
## Neutrophil:uropathogenic_E._coli_UTI89
## 1
## Osteoblasts
## 9
## Osteoblasts:BMP2
## 6
## Platelets
## 5
## Pre-B_cell_CD34-
## 2
## Pro-B_cell_CD34+
## 2
## Pro-Myelocyte
## 2
## Smooth_muscle_cells:bronchial
## 3
## Smooth_muscle_cells:bronchial:vit_D
## 3
## Smooth_muscle_cells:umbilical_vein
## 2
## Smooth_muscle_cells:vascular
## 5
## Smooth_muscle_cells:vascular:IL-17
## 3
## T_cell:CCR10+CLA+1,25(OH)2_vit_D3/IL-12
## 1
## T_cell:CCR10-CLA+1,25(OH)2_vit_D3/IL-12
## 1
## T_cell:CD4+
## 12
## T_cell:CD4+_Naive
## 6
## T_cell:CD4+_central_memory
## 5
## T_cell:CD4+_effector_memory
## 4
## T_cell:CD8+
## 16
## T_cell:CD8+_Central_memory
## 3
## T_cell:CD8+_effector_memory
## 4
## T_cell:CD8+_effector_memory_RA
## 4
## T_cell:CD8+_naive
## 4
## T_cell:Treg:Naive
## 2
## T_cell:effector
## 4
## T_cell:gamma-delta
## 2
## Tissue_stem_cells:BM_MSC
## 8
## Tissue_stem_cells:BM_MSC:BMP2
## 12
## Tissue_stem_cells:BM_MSC:TGFb3
## 11
## Tissue_stem_cells:BM_MSC:osteogenic
## 8
## Tissue_stem_cells:CD326-CD56+
## 3
## Tissue_stem_cells:adipose-derived_MSC_AM3
## 2
## Tissue_stem_cells:dental_pulp
## 6
## Tissue_stem_cells:iliac_MSC
## 3
## Tissue_stem_cells:lipoma-derived_MSC
## 2
## iPS_cells:CRL2097_foreskin
## 3
## iPS_cells:CRL2097_foreskin-derived:d20_hepatic_diff
## 3
## iPS_cells:CRL2097_foreskin-derived:undiff.
## 3
## iPS_cells:PDB_1lox-17Puro-10
## 1
## iPS_cells:PDB_1lox-17Puro-5
## 1
## iPS_cells:PDB_1lox-21Puro-20
## 1
## iPS_cells:PDB_1lox-21Puro-26
## 1
## iPS_cells:PDB_2lox-17
## 1
## iPS_cells:PDB_2lox-21
## 1
## iPS_cells:PDB_2lox-22
## 1
## iPS_cells:PDB_2lox-5
## 1
## iPS_cells:PDB_fibroblasts
## 1
## iPS_cells:adipose_stem_cell-derived:lentiviral
## 3
## iPS_cells:adipose_stem_cell-derived:minicircle-derived
## 3
## iPS_cells:adipose_stem_cells
## 3
## iPS_cells:fibroblast-derived:Direct_del._reprog
## 2
## iPS_cells:fibroblast-derived:Retroviral_transf
## 1
## iPS_cells:fibroblasts
## 1
## iPS_cells:foreskin_fibrobasts
## 1
## iPS_cells:iPS:minicircle-derived
## 5
## iPS_cells:skin_fibroblast
## 2
## iPS_cells:skin_fibroblast-derived
## 3
table(blueprint_encode$main_types)
##
## Adipocytes B-cells CD4+ T-cells CD8+ T-cells
## 11 8 14 5
## Chondrocytes DC Endothelial cells Eosinophils
## 2 1 26 1
## Epithelial cells Erythrocytes Fibroblasts HSC
## 18 7 20 38
## Keratinocytes Macrophages Melanocytes Mesangial cells
## 2 25 4 2
## Monocytes Myocytes NK cells Neurons
## 16 4 3 4
## Neutrophils Pericytes Skeletal muscle Smooth muscle
## 23 2 7 16
table(blueprint_encode$types)
##
## Adipocytes Astrocytes
## 7 2
## CD4+ T-cells CD4+ Tcm
## 11 1
## CD4+ Tem CD8+ T-cells
## 1 3
## CD8+ Tcm CD8+ Tem
## 1 1
## CLP CMP
## 5 11
## Chondrocytes Class-switched memory B-cells
## 2 1
## DC Endothelial cells
## 1 18
## Eosinophils Epithelial cells
## 1 18
## Erythrocytes Fibroblasts
## 7 20
## GMP HSC
## 3 6
## Keratinocytes MEP
## 2 4
## MPP Macrophages
## 4 18
## Macrophages M1 Macrophages M2
## 3 4
## Megakaryocytes Melanocytes
## 5 4
## Memory B-cells Mesangial cells
## 1 2
## Monocytes Myocytes
## 16 4
## NK cells Neurons
## 3 4
## Neutrophils Pericytes
## 23 2
## Plasma cells Preadipocytes
## 4 2
## Skeletal muscle Smooth muscle
## 7 16
## Tregs mv Endothelial cells
## 1 8
## naive B-cells
## 2
Using SingleR with other reference data sets
SingleR only needs a single gene expression profile per cell type, which makes it possible to use bulk-RNAsequencing and even micorarrays as reference data. The downside is that the variability within cell types is not represented (although their methods do provide a p-value. Again, see SingleR’s highly recommend vignettes).
The other method we will look at, CHETAH, needs several (100-200) single-cell expression profiles for the classification. The advantage is that the inherent variability is fully account for. CHETAH, originally developed for working with cancer data has its ‘own’ reference data set
that is based on single-cell data from Head-Neck cancer, melanoma, breast and colorectal cancer. (For details see https://figshare.com/s/aaf026376912366f81b6) Note that it is easy to create your own reference data sets for both SingleR and CHETAH.
SingleR can use the CHETAH reference if that has been ‘bulkified’ by
averaging over all cells per reference cell type. We provide this as a
ready-made object (chetah.ref.singler).
The layout of the reference data is quite simple: a list with the name
of the reference, a big data matrix (genes x celltypes), and the types
per cell, both in a detailed version ($types) and the simple version
($main_types) For the CHETAH reference we duplicated the ($types)
and the simple version ($main_types). (Note that the
chetah.ref.singler reference object can only be used by SingleR, not
by CHETAH).
file <- paste0(data.dir, "/chetah.ref.singler.rds")
chetah.ref.singler <- readRDS(file=file)
# which main type are there:
unique(chetah.ref.singler$main_types)
## [1] "B cell" "Macrophage" "NK" "CAF"
## [5] "Myofibroblast" "Endothelial" "CD4 T cell" "CD8 T cell"
## [9] "reg. T cell" "Mast" "Dendritic" "Plasma"
# layout of the object:
str(chetah.ref.singler)
## List of 4
## $ name : chr "CHETAH reference"
## $ data : num [1:18588, 1:12] 0.6527 0.0007 0.2441 0.0258 0.6127 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:18588] "ELMO2" "CREB3L1" "PNMA1" "MMP2" ...
## .. ..$ : chr [1:12] "B cell" "Macrophage" "NK" "CAF" ...
## $ types : chr [1:12] "B cell" "Macrophage" "NK" "CAF" ...
## $ main_types: chr [1:12] "B cell" "Macrophage" "NK" "CAF" ...
Classifying with SingleR
SingleR can classify using several different reference data sets at the same time; this saves time and memory.
On to the actual classification with SingleR.
counts <- GetAssayData(ovarian)
singler <- CreateSinglerObject(counts=counts,
project.name="excelerate course", # choose
min.genes = 200, # ignore cells with fewer than 200 transcripts
technology = "CEL-Seq2", # choose
species = "Human",
citation = "Schelker et al. 2017", # choose
ref.list = list(hpca=hpca, bpe=blueprint_encode, snglr_chetah=chetah.ref.singler),
normalize.gene.length = FALSE, # needed for full-length platforms (e.g. smartseq)
variable.genes = "de", # see vignette
fine.tune = FALSE, # TRUE would take very long
reduce.file.size = TRUE, # leave out less-often used fields
do.signatures = FALSE,
do.main.types = TRUE,
numCores = SingleR.numCores)
## [1] "Dimensions of counts data: 25462x1200"
## [1] "Annotating data with HPCA..."
## [1] "Variable genes method: de"
## [1] "Number of DE genes:4401"
## [1] "Number of cells: 1200"
## [1] "Number of DE genes:4401"
## [1] "Number of clusters: 10"
## [1] "Annotating data with HPCA (Main types)..."
## [1] "Number of DE genes:3312"
## [1] "Number of cells: 1200"
## [1] "Number of DE genes:3312"
## [1] "Number of clusters: 10"
## [1] "Annotating data with Blueprint_Encode..."
## [1] "Variable genes method: de"
## [1] "Number of DE genes:3756"
## [1] "Number of cells: 1200"
## [1] "Number of DE genes:3756"
## [1] "Number of clusters: 10"
## [1] "Annotating data with Blueprint_Encode (Main types)..."
## [1] "Number of DE genes:3183"
## [1] "Number of cells: 1200"
## [1] "Number of DE genes:3183"
## [1] "Number of clusters: 10"
## [1] "Annotating data with CHETAH reference..."
## [1] "Variable genes method: de"
## [1] "Number of DE genes:1682"
## [1] "Number of cells: 1200"
## [1] "Number of DE genes:1682"
## [1] "Number of clusters: 10"
## [1] "Annotating data with CHETAH reference (Main types)..."
## [1] "Number of DE genes:1682"
## [1] "Number of cells: 1200"
## [1] "Number of DE genes:1682"
## [1] "Number of clusters: 10"
The ref.list argument specified a named list with three different
reference data sets: HPCA, blueprint_encode (‘bpe’) and the bulkified
chetah_reference (‘snglr_chetah’). The resulting singler object has
complete classifications for each of these reference sets, under the
$singler member. The actual types per cell are found in sub-list
$SingleR.single.main$labels[,1].
(side-note: SingleR also automatically classifies per cluster of cells, but we will not use this type of classification.)
To get a good overview it’s easiest to iterate over all elements of this list.
show(names(singler$singler))
## [1] "hpca" "bpe" "snglr_chetah"
for (ref.set in names(singler$singler) ) {
types <- singler$singler[[ref.set]]$SingleR.single.main$labels[,1]
cat("==== ", ref.set, ": ====\n")
show(sort(table(types), decreasing=TRUE))
}
## ==== hpca : ====
## types
## Monocyte Macrophage T_cells
## 528 277 142
## Epithelial_cells NK_cell Pre-B_cell_CD34-
## 109 42 39
## DC B_cell Smooth_muscle_cells
## 18 9 9
## Fibroblasts HSC_-G-CSF Embryonic_stem_cells
## 7 5 3
## Neutrophils GMP Neurons
## 3 2 2
## iPS_cells MEP Platelets
## 2 1 1
## Pro-B_cell_CD34+
## 1
## ==== bpe : ====
## types
## Macrophages Monocytes CD4+ T-cells Epithelial cells
## 521 301 118 107
## B-cells CD8+ T-cells NK cells DC
## 50 41 28 9
## Mesangial cells Fibroblasts Adipocytes Eosinophils
## 8 6 4 2
## Pericytes Endothelial cells HSC Neutrophils
## 2 1 1 1
## ==== snglr_chetah : ====
## types
## Macrophage CD4 T cell Endothelial B cell NK
## 830 139 94 42 28
## CAF CD8 T cell Dendritic Myofibroblast Plasma
## 20 20 15 9 3
## For hpca and blueprint_encode also show the
## detailed cell typings (as opposed to main_types results) :
for (ref.set in c("hpca", "bpe") ) {
types <- singler$singler[[ref.set]]$SingleR.single$labels[,1]
subrefset <- paste(ref.set, "subtypes", sep="_")
cat("==== ", subrefset, ": ====\n")
show(sort(table(types), decreasing=TRUE))
}
## ==== hpca_subtypes : ====
## types
## Monocyte:leukotriene_D4
## 197
## Macrophage:monocyte-derived:M-CSF
## 144
## Macrophage:monocyte-derived:IL-4/Dex/TGFb
## 112
## Macrophage:monocyte-derived:IL-4/Dex/cntrl
## 109
## T_cell:CD4+_central_memory
## 95
## Monocyte:CD16-
## 92
## Epithelial_cells:bladder
## 57
## Epithelial_cells:bronchial
## 47
## Monocyte:CD14+
## 36
## Pre-B_cell_CD34-
## 34
## T_cell:CD4+_effector_memory
## 29
## Macrophage:monocyte-derived:M-CSF/IFNg
## 28
## DC:monocyte-derived:AEC-conditioned
## 25
## Monocyte:anti-FcgRIIB
## 23
## NK_cell
## 22
## Monocyte:CD16+
## 19
## NK_cell:IL2
## 15
## T_cell:CD4+
## 11
## Macrophage:monocyte-derived
## 9
## Monocyte
## 8
## Monocyte:S._typhimurium_flagellin
## 7
## T_cell:CD4+_Naive
## 7
## B_cell:immature
## 6
## Smooth_muscle_cells:vascular:IL-17
## 6
## DC:monocyte-derived:mature
## 4
## Macrophage:monocyte-derived:M-CSF/Pam3Cys
## 4
## NK_cell:CD56hiCD62L+
## 4
## Smooth_muscle_cells:vascular
## 4
## T_cell:CD8+
## 4
## B_cell:Naive
## 3
## DC:monocyte-derived:AM580
## 3
## Fibroblasts:breast
## 3
## Macrophage:monocyte-derived:IL-4/TGFb
## 3
## Neutrophil:commensal_E._coli_MG1655
## 3
## iPS_cells:iPS:minicircle-derived
## 3
## B_cell:Memory
## 2
## DC:monocyte-derived:anti-DC-SIGN_2h
## 2
## Endothelial_cells:lymphatic:TNFa_48h
## 2
## HSC_-G-CSF
## 2
## T_cell:Treg:Naive
## 2
## B_cell:CXCR4-_centrocyte
## 1
## B_cell:Plasma_cell
## 1
## DC:monocyte-derived
## 1
## DC:monocyte-derived:antiCD40/VAF347
## 1
## GMP
## 1
## MSC
## 1
## Macrophage:Alveolar
## 1
## Macrophage:monocyte-derived:M-CSF/IFNg/Pam3Cys
## 1
## Macrophage:monocyte-derived:S._aureus
## 1
## Monocyte:CXCL4
## 1
## Osteoblasts:BMP2
## 1
## T_cell:gamma-delta
## 1
## iPS_cells:CRL2097_foreskin-derived:undiff.
## 1
## iPS_cells:PDB_1lox-21Puro-26
## 1
## ==== bpe_subtypes : ====
## types
## Macrophages Monocytes
## 567 249
## Epithelial cells CD4+ T-cells
## 101 42
## CD4+ Tcm Memory B-cells
## 41 34
## CD4+ Tem NK cells
## 31 26
## CD8+ Tem CD8+ Tcm
## 21 13
## Mesangial cells Tregs
## 13 12
## DC Adipocytes
## 10 7
## naive B-cells Fibroblasts
## 6 5
## Macrophages M1 Plasma cells
## 4 4
## Class-switched memory B-cells CLP
## 3 2
## Endothelial cells HSC
## 2 2
## CD8+ T-cells Chondrocytes
## 1 1
## Eosinophils Macrophages M2
## 1 1
## Neutrophils
## 1
We will stick to the main_types from now on, for brevity. In order to
easily visualize the various classifications in the tSNE plots we have
to add them to meta.data slot of the Seurat object:
for (ref.set in names(singler$singler) ) {
types <- singler$singler[[ref.set]]$SingleR.single.main$labels[,1]
ovarian <- AddMetaData(ovarian,
metadata=types,
col.name=paste0(ref.set,"_type" ) )
}
## Check if this worked and get an impression of the concordance of classification
interesting.columns <- c("celltypes", "hpca_type", "bpe_type", "snglr_chetah_type")
## repeat the following a few times:
random.rows <- sort(sample(ncol(ovarian), size=20))
ovarian@meta.data[ random.rows, interesting.columns]
## celltypes hpca_type bpe_type snglr_chetah_type
## X7873M.31 Macrophage Macrophage Macrophages Macrophage
## X7873M.413 Macrophage Monocyte Macrophages Macrophage
## X7873M.455 Macrophage Monocyte Monocytes Macrophage
## X7873M.462 Macrophage Macrophage Macrophages Macrophage
## X7873M.785 Macrophage Macrophage Macrophages Macrophage
## X7882M.547 Macrophage Monocyte Macrophages Macrophage
## X7882M.550 Dendritic Pre-B_cell_CD34- B-cells B cell
## X7882M.582 Unknown NK_cell CD4+ T-cells CD4 T cell
## X7882M.746 Macrophage Monocyte Monocytes Macrophage
## X7882M.838 Macrophage Monocyte Monocytes Macrophage
## X7892M.300 Macrophage Monocyte Macrophages Macrophage
## X7892M.447 Macrophage Macrophage Macrophages Macrophage
## X7892M.482 Macrophage Macrophage Macrophages Macrophage
## X7892M.545 Macrophage Monocyte Macrophages Macrophage
## X7990M2.245 Tumor Epithelial_cells Epithelial cells Endothelial
## X7990M2.34 Macrophage Macrophage Macrophages Macrophage
## X7990M2.343 Tumor Epithelial_cells Epithelial cells Endothelial
## X7990M2.455 Macrophage Monocyte Monocytes Macrophage
## X7990M2.466 Macrophage Monocyte Macrophages Macrophage
## X7990M2.82 CD4 T cell T_cells CD4+ T-cells CD4 T cell
Things seem largely concordant, now start plotting them.
panel.labels <- c('publ','hpca','bpe','chet') #shorthand labels
p1 <- DimPlot(ovarian, group.by="celltypes", label_size=6, reduction=dim.red)
p2 <- DimPlot(ovarian, group.by='hpca_type', label_size=6, reduction=dim.red)
p3 <- DimPlot(ovarian, group.by='bpe_type', label_size=6, reduction=dim.red)
p4 <- DimPlot(ovarian, group.by='snglr_chetah_type', label_size=6, reduction=dim.red)
plot_grid(p1, p2, p3, p4, nrow=2, ncol=2, labels=panel.labels)

This looks reasonable, but the colors are a bit messy. To see things
better it may be better to highlight the cell type of interest (but note
that the type names differ per reference set!!). The Seurat function
WhichCells is a bit too limited to find cells by ‘any old meta data’,
so it’s easier to use a little function to automatically find the cells
that have a particular type:
findCells <- function(obj, column, values, name=NULL) {
## Given a Seurat OBJ, return a list with the names of the cells where
## the specified meta.data COLUMN equals any of the strings specified
## in VALUES (both must be characters or factors). Name of the list member
## must be specified using the NAME argument if length(values)>1
stopifnot(is(obj, "Seurat"))
stopifnot(is.character(column))
stopifnot(column %in% names(obj@meta.data))
col <- obj@meta.data[[column]]
stopifnot(is.character(col) || is.factor(col))
values <- unique(values)
stopifnot(is.character(values) || is.factor(values))
if (length(values)>1 && is.null(name))
stop("findCells: specify a name to be used for the selection")
if(is.null(name))
name <- values
stopifnot(is.character(name))
rem <- setdiff(c(values), col)
if(length(rem)>0)stop("findCells: requested value(s) never occurs in this column: ", rem)
l <- list(colnames(obj)[ col %in% values ])
names(l) <- name
l
} #findCells
## "Let's look at the macrophages. This is the biggest group
p1 <- DimPlot(ovarian, group.by="celltypes", reduction=dim.red,
cells.highlight=findCells(ovarian, 'celltypes', 'Macrophage'))
p2 <- DimPlot(ovarian, group.by='hpca_type', reduction=dim.red,
cells.highlight=findCells(ovarian, 'hpca_type', 'Macrophage'))
p3 <- DimPlot(ovarian, group.by='bpe_type', reduction=dim.red,
cells.highlight=findCells(ovarian, 'bpe_type', 'Macrophages'))
p4 <- DimPlot(ovarian, group.by='snglr_chetah_type', reduction=dim.red,
cells.highlight=findCells(ovarian, 'snglr_chetah_type', 'Macrophage'))
plot_grid(p1, p2, p3, p4, nrow=2, ncol=2, labels=paste(panel.labels, "Macrophage"))
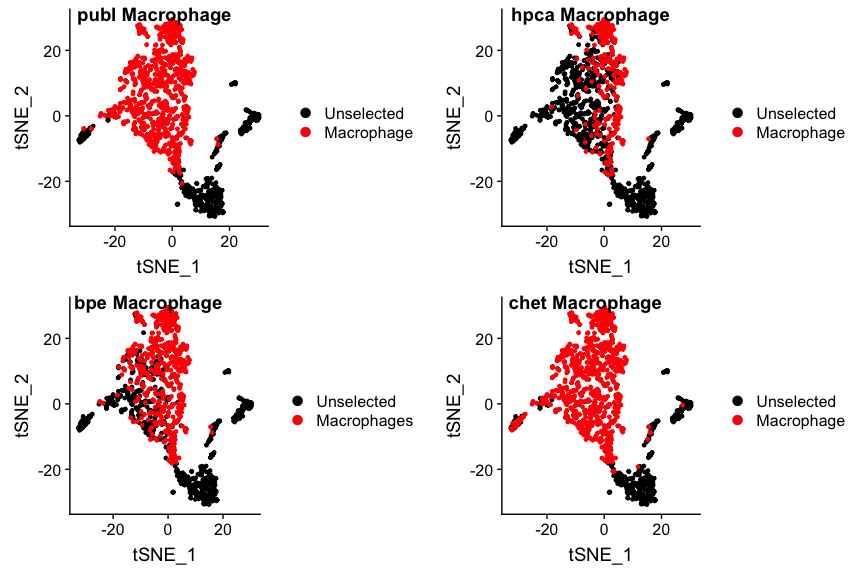
No suprises there really. Note that monocytes (found by HPCA) are
precursors to macrophages, but are called differently. That’s why there
are relatively fewer macrophages in the hpca plot.
This ‘missing-things-that-are-too-specific’ is more prominent for the B cells as identified by SingleR using the four different references:
p1 <- DimPlot(ovarian, group.by="celltypes", reduction=dim.red,
cells.highlight=findCells(ovarian, 'celltypes', 'B cell'))
p2 <- DimPlot(ovarian, group.by='hpca_type', reduction=dim.red,
cells.highlight=findCells(ovarian, 'hpca_type', 'B_cell'))
p3 <- DimPlot(ovarian, group.by='bpe_type', reduction=dim.red,
cells.highlight=findCells(ovarian, 'bpe_type', 'B-cells'))
p4 <- DimPlot(ovarian, group.by='snglr_chetah_type', reduction=dim.red,
cells.highlight=findCells(ovarian, 'snglr_chetah_type', 'B cell'))
plot_grid(p1, p2, p3, p4, nrow=2, ncol=2, labels=paste(panel.labels, "B cell"))
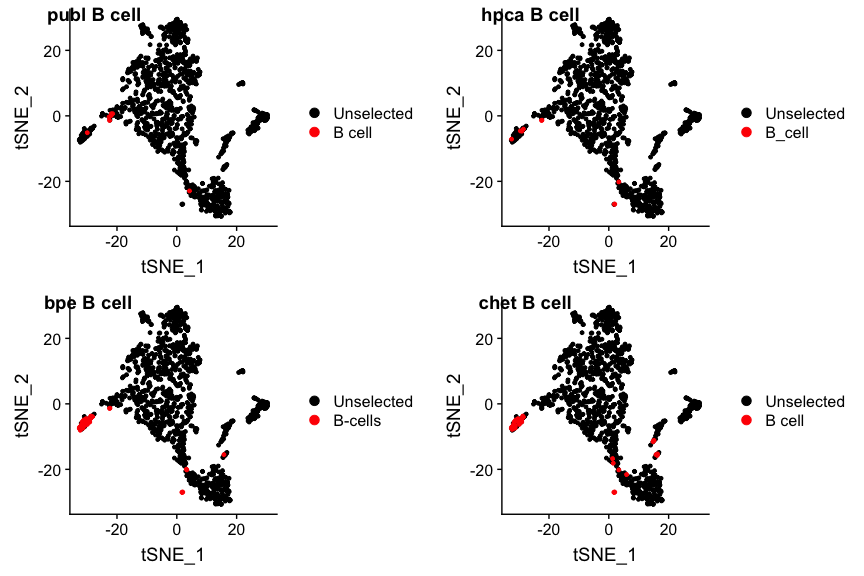
bpe and singlr_chetah find more B-cells than the original publication,
HPCA roughly the same. The reason is that HPCA has two more B-cell
subtypes which were missed. We can lump them together (findCells() can
handle it) by including the ‘Pre-B_cell_CD34-’ and
‘Pro-B_cell_CD34+’ cells, and calling the combination ‘B-like’ :
p2 <- DimPlot(ovarian, group.by='hpca_type', reduction=dim.red,
cells.highlight=findCells(ovarian, 'hpca_type',
c('B_cell', 'Pre-B_cell_CD34-', 'Pro-B_cell_CD34+'),
name="B-like"))
plot_grid(p1, p2, p3, p4, nrow=2, ncol=2, labels=paste(panel.labels, "all B cell"))
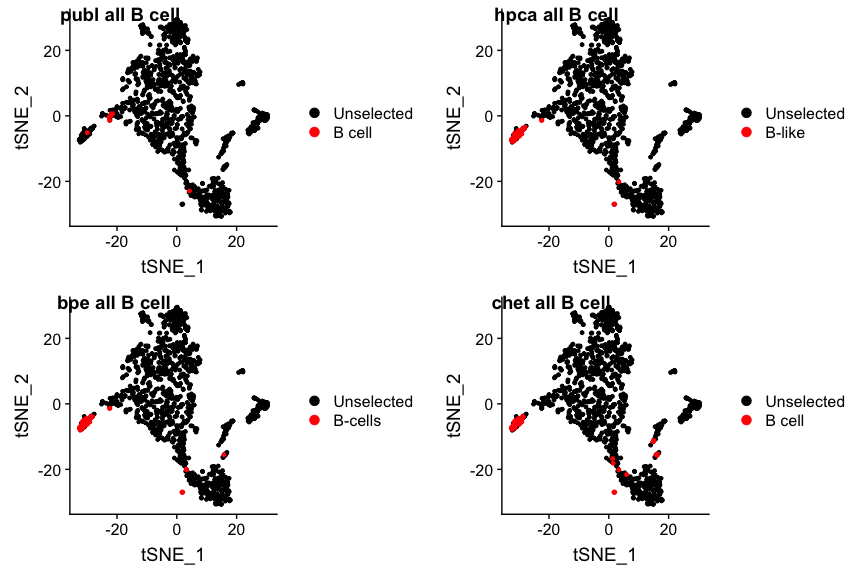
It looks like in the original publication quite a few B(-like) cells were missed. The opposite may be the case for the dendritic cells: the publication assigns quite a few of them, which is a bit unusual in that these tissue-residing cells are relatively rare in fluids such as blood and ascites. Are the authors right?
p1 <- DimPlot(ovarian, group.by="celltypes", reduction=dim.red,
cells.highlight=findCells(ovarian, 'celltypes', 'Dendritic'))
p2 <- DimPlot(ovarian, group.by='hpca_type', reduction=dim.red,
cells.highlight=findCells(ovarian, 'hpca_type', 'DC'))
p3 <- DimPlot(ovarian, group.by='bpe_type', reduction=dim.red,
cells.highlight=findCells(ovarian, 'bpe_type', 'DC'))
p4 <- DimPlot(ovarian, group.by='snglr_chetah_type', reduction=dim.red,
cells.highlight=findCells(ovarian, 'snglr_chetah_type', 'Dendritic'))
plot_grid(p1, p2, p3, p4, nrow=2, ncol=2, labels=paste(panel.labels, "Dendritic"))
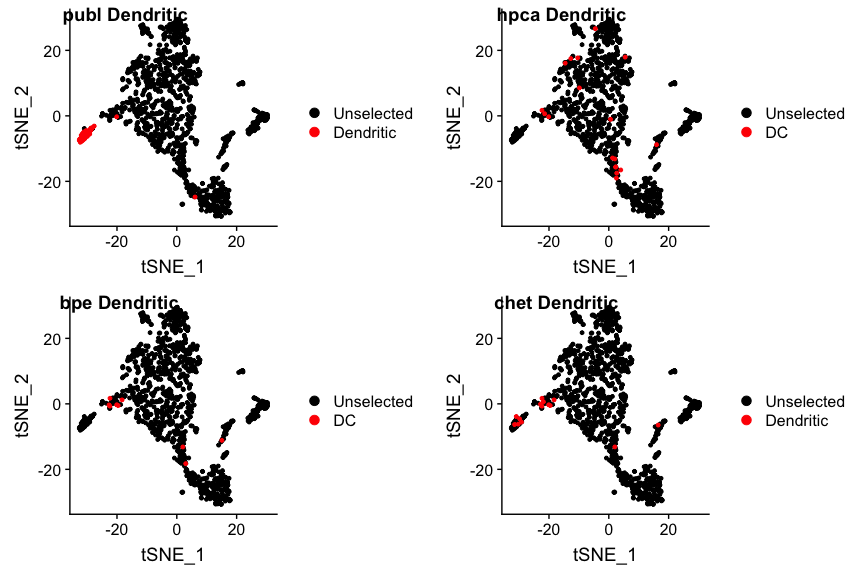
I guess the jury is out, but more on that later.
Feel free to play around with a few more cell types. In particular, have a good look at where the tumor cells are. You will find that there are 4 clusters. I’m pretty sure each cluster derive from a different patient.
Discrepancies
A nice way to view any discrepancies is to split the cells by one
classification, and color them by another (and perhaps vice versa). If
there are no discrepancies, each sub-plot has cells of one color. (you
can try this by using the same cell typing for both the split.by and
group.by arguments).
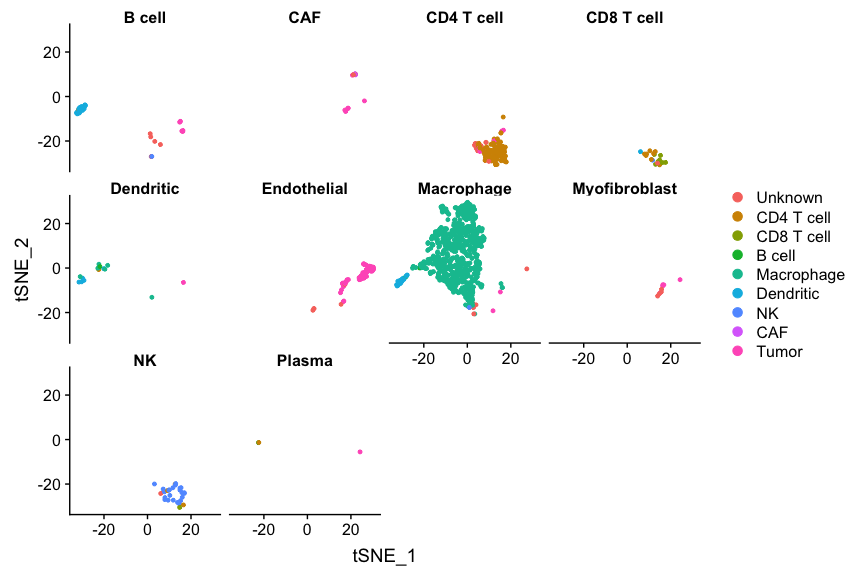
DimPlot(ovarian, split.by='celltypes', group.by='snglr_chetah_type', reduction=dim.red)
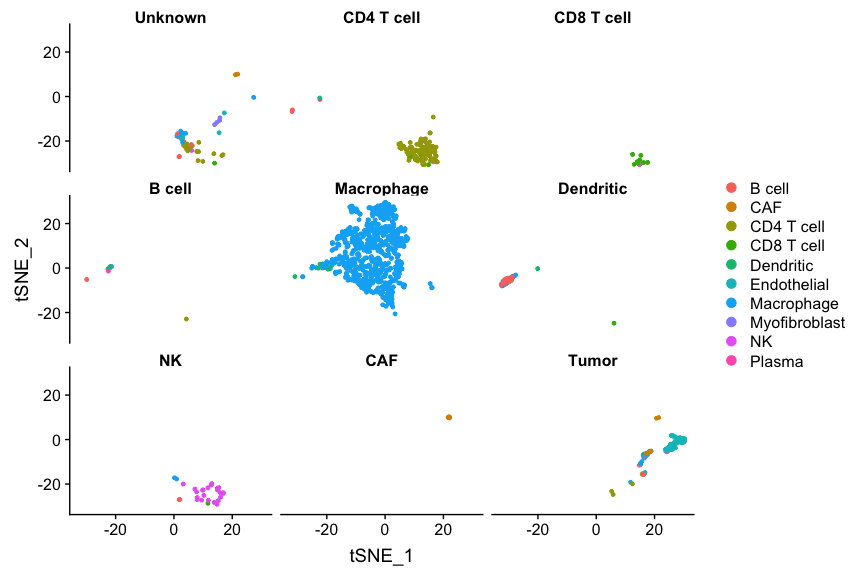
## and also in reverse:
DimPlot(ovarian, group.by='celltypes', split.by='snglr_chetah_type', reduction=dim.red)
CHETAH
CHETAH reference
Our second method, CHETAH, differs from SingleR in that it uses a reference in which each cell type is represented by a few hundred single-cells of that type, allowing a well-founded estimate of the confidence with which a cell type call can be made. We already worked with a ‘bulkified’ version of this reference in the previous section.
CHETAH is part of Bioconductor, and therefore uses
SingleCellExperiment objects, both for the data to be classified (the
‘input’) as well as for the reference data. The latter is simply a
data set that has celltypes as one of its colData() columns.
Let’s look at this reference.
file <- paste0(data.dir,"/chetah.ref.rds")
chetah.ref <- readRDS(file=file)
show(unique(chetah.ref.singler$types)) # the bulkified reference we've used
## [1] "B cell" "Macrophage" "NK" "CAF"
## [5] "Myofibroblast" "Endothelial" "CD4 T cell" "CD8 T cell"
## [9] "reg. T cell" "Mast" "Dendritic" "Plasma"
show(colData(chetah.ref)) # the CHETAH tumor reference data
## DataFrame with 6122 rows and 1 column
## celltypes
## <character>
## HN25_P24_B12_S120_comb B cell
## HN25_P5_E03_S51_comb B cell
## HNSCC26_P24_A06_S294_comb B cell
## HNSCC25_P2_H06_S90_comb B cell
## HNSCC28_P12_A04_S196_comb B cell
## ... ...
## CY88CD45POS_2_H10_S478_comb Plasma
## CY94CD45POS_1_H04_S184_comb Plasma
## CY88CD45POS_7_C05_S221_comb Plasma
## CY94_CD45NEG_CD90POS_2_B04_S16_comb Plasma
## CY94_CD45NEG_CD90POS_2_E09_S57_comb Plasma
show(sort(table(colData(chetah.ref)$celltypes), decreasing=TRUE))
##
## CD8 T cell CD4 T cell B cell Myofibroblast CAF
## 1643 1044 769 693 685
## Endothelial Macrophage reg. T cell Plasma Mast
## 360 225 225 164 126
## Dendritic NK
## 101 87
Classifying with SingleR
The data to be classified also must be cast as a SingleCellExperiment.
You can use Seurat’s as.SingleCellExperiment() function for that.
Classifying is a matter of calling CHETAHclassifier() with the input
and the reference as arguments (although there are loads of options, see
the man page). Classification takes a bit longer than SingleR
ovarian.sce <- as.SingleCellExperiment(ovarian)
ovarian.sce <- CHETAHclassifier(input=ovarian.sce,
ref_cells = chetah.ref)
## Preparing data....
## Running analysis...
Let’s explore the results. You’ll notice an extra column
celltype_CHETAH in the colData, and things are still by and large
concordant, with one exception: CHETAH uses the odd ‘Unassigned’ and
‘Node3’, ‘Node7’ etc. type. The largest group, as expected, is again
Macrophage
(Note: the snglr_chetah_type column is the SingleR classification
using the bulkified CHETAH reference, whereas the celltype_CHETAH
column is the CHETAH classifcation using the CHETAH reference.)
names(colData(ovarian.sce))
## [1] "nGene" "nUMI" "orig.ident"
## [4] "celltypes" "nCount_RNA" "nFeature_RNA"
## [7] "RNA_snn_res.0.5" "seurat_clusters" "hpca_type"
## [10] "bpe_type" "snglr_chetah_type" "ident"
## [13] "celltype_CHETAH"
interesting.columns <- c("celltypes", "hpca_type", "bpe_type", "snglr_chetah_type", "celltype_CHETAH")
## repeat the following a few times
random.rows <- sort(sample(ncol(ovarian), size=20))
colData(ovarian.sce)[ random.rows, interesting.columns]
## DataFrame with 20 rows and 5 columns
## celltypes hpca_type bpe_type snglr_chetah_type
## <factor> <character> <character> <character>
## X7873M.110 Macrophage Monocyte Macrophages Macrophage
## X7873M.121 Macrophage Monocyte Monocytes Macrophage
## X7873M.213 Macrophage Macrophage Macrophages Macrophage
## X7873M.643 Macrophage Macrophage Macrophages Macrophage
## X7882M.285 Macrophage Monocyte Monocytes Macrophage
## ... ... ... ... ...
## X7892M.774 Macrophage Monocyte Monocytes Macrophage
## X7892M.84 Macrophage Macrophage Macrophages Macrophage
## X7990M2.258 Unknown T_cells CD4+ T-cells CD4 T cell
## X7990M2.280 Macrophage Monocyte Macrophages Macrophage
## X7990M2.42 NK B_cell B-cells B cell
## celltype_CHETAH
## <character>
## X7873M.110 Macrophage
## X7873M.121 Macrophage
## X7873M.213 Macrophage
## X7873M.643 Unassigned
## X7882M.285 Macrophage
## ... ...
## X7892M.774 Macrophage
## X7892M.84 Macrophage
## X7990M2.258 Unassigned
## X7990M2.280 Macrophage
## X7990M2.42 B cell
show(sort(table(colData(ovarian.sce)$celltype_CHETAH), decreasing=TRUE))
##
## Macrophage Unassigned CD4 T cell Dendritic Node9 Node5
## 706 204 90 34 32 27
## Node7 Node8 NK CD8 T cell Node1 Node2
## 27 22 19 15 12 6
## B cell Node6 Node3 Plasma
## 2 2 1 1
CHETAH visualization
Since the CHETAH method is so inherenty dependent on the classification tree, it has its own routine to visualize both at the same time.
## There may still be a small bug in CHETAH: if things don't work,
## please use the following code before continuing
pca.save <- ovarian.sce@reducedDims@listData$PCA
ovarian.sce@reducedDims@listData$PCA <- NULL
ovarian.sce@reducedDims@listData$PCA <- pca.save[,1:2]
## end of workaround
dim.red.u <- toupper(dim.red) # CHETAH uses upper case TSNE, UMAP, etc.
PlotCHETAH(ovarian.sce, redD=dim.red.u)
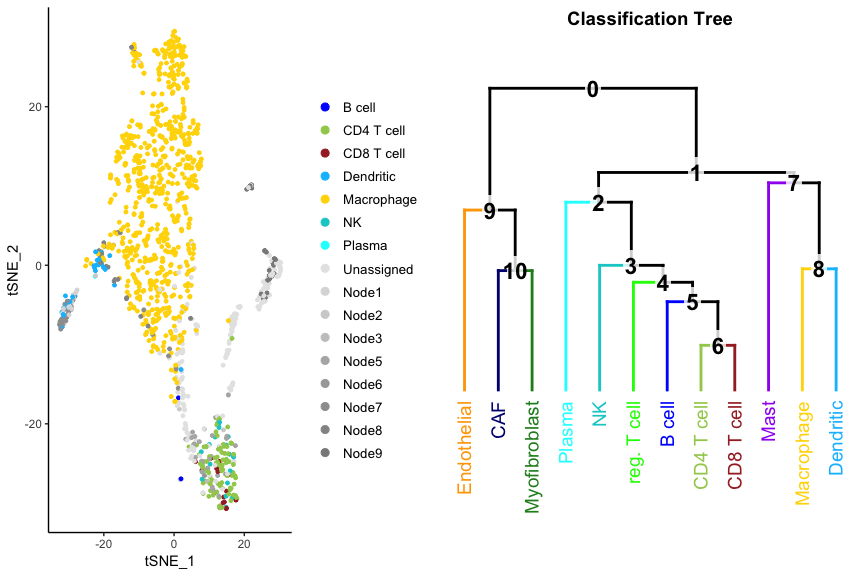
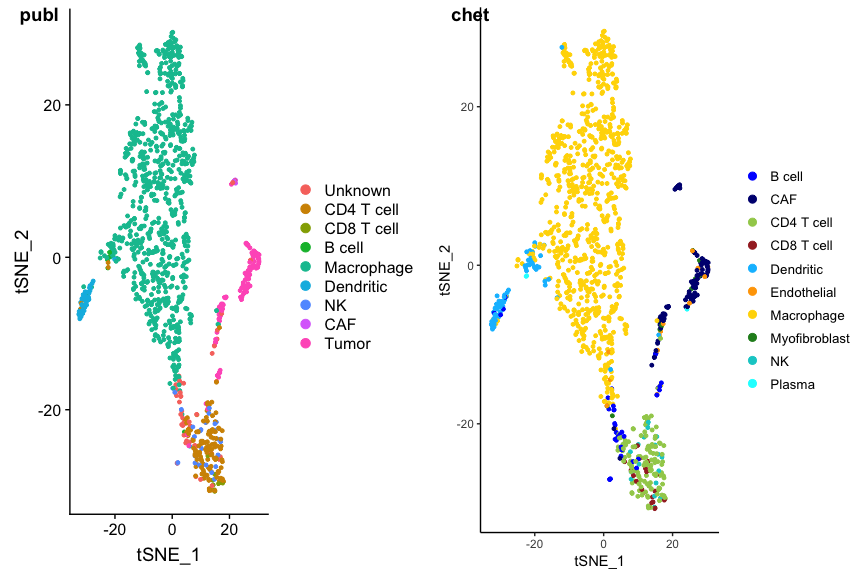
Grey dots are cells that got an intermediate classification, here called Unassigned, Node1, Node2 etc. You can immediately see the four clusters that could not be classified: they are the tumor cells. A check with the author classification confirms this:
p1 <- DimPlot(ovarian, group.by='celltypes', reduction=dim.red)
p2 <- PlotCHETAH(ovarian.sce, redD=dim.red.u, tree=FALSE, return=TRUE)
plot_grid(p1, p2, ncol=2, labels=c("publ", "chet"))
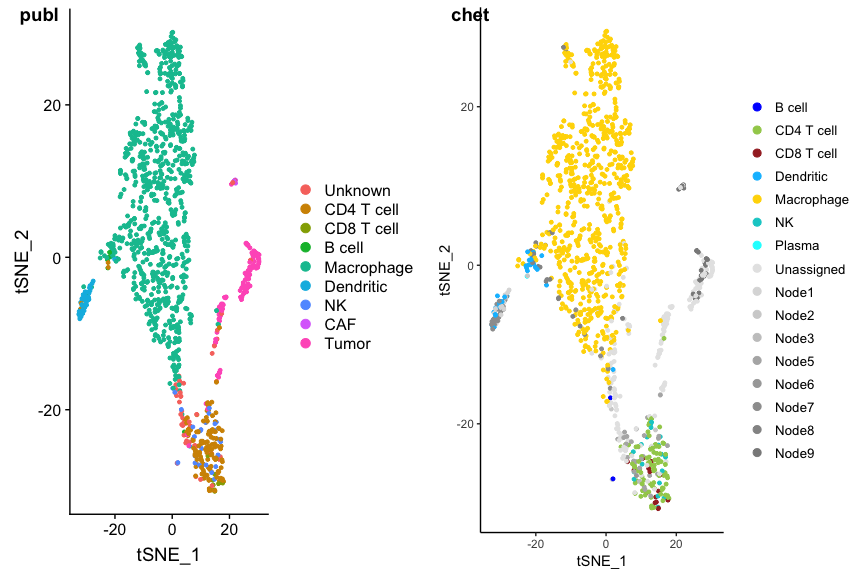
SingleR doesn’t know CHETAH’s intermediate types, so using SingleR with CHETAH’s reference gives different (and misleading) results:
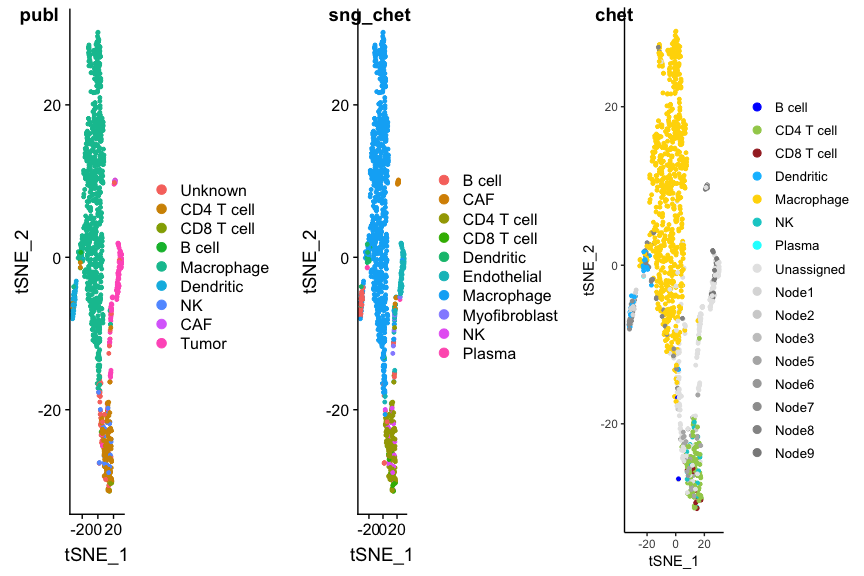
p1 <- DimPlot(ovarian, group.by='celltypes', reduction=tolower(dim.red))
p2 <- DimPlot(ovarian, group.by="snglr_chetah_type", reduction=tolower(dim.red))
p3 <- PlotCHETAH(ovarian.sce, redD=dim.red.u, tree=FALSE, return=TRUE)
plot_grid(p1, p2, p3, ncol=3, labels=c("publ", "sng_chet", "chet"))
To see the details of the intermediate classifications more clearly you
can invert the color scheme using the interm option. The malignant
cells stand out even more clearly this way.
p2 <- PlotCHETAH(ovarian.sce, redD=dim.red.u, interm=TRUE, tree=FALSE, return=TRUE)
plot_grid(p1, p2, ncol=2, labels=c("publ", "chet"))
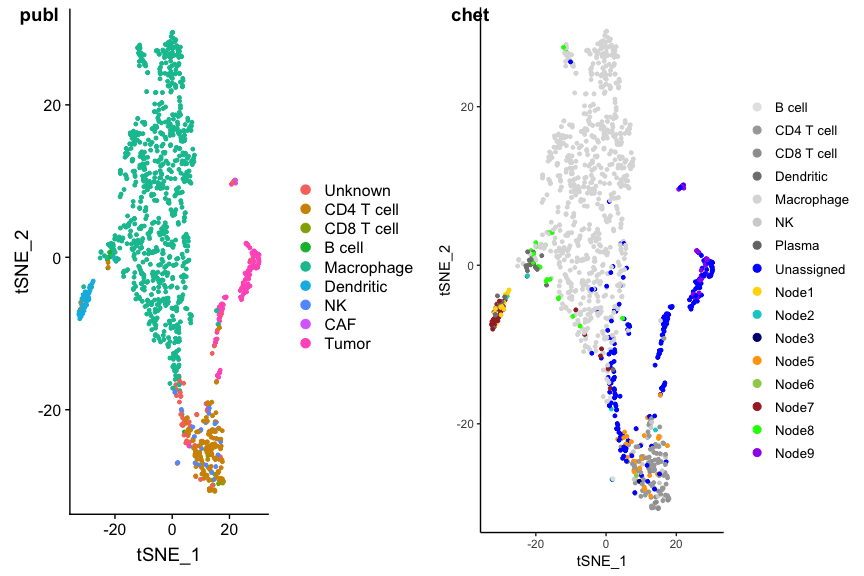
The dendritic cell type calls also look a bit more suspect. We can play
with the classification threshold (the default is 0.1) By setting it to
0, we force all cells to be classified to a final type; no intermediates
will occur then. Adjusting the threshold parameter is done with the
Classify function (this is very fast). Play with it. E.g., you’ll see
that the dendritic calls from the original publication may be in fact be
plausible but were missed by SingleR. (But also keep track of what
happens with the types of the tumor cells!)
threshold <- 0.0
ovarian.sce <- Classify(ovarian.sce, thresh=threshold)
## note: this overrides previous celltype_CHETAH, but is very fast anyway
p2 <- PlotCHETAH(ovarian.sce, redD=dim.red.u, interm=FALSE, tree=FALSE, return=TRUE)
plot_grid(p1, p2, ncol=2, labels=c("publ", "chet"))
CHETAHshiny
CHETAH comes with a nice Shiny app that makes it easy to explore the classification. It makes R start a little web application that you can interact with in your web browser.
Launching
When calling CHETAHshiny(), it should say something like “Listening on
http://127.0.0.1:5433” and automatically launch your web browser. If
not, manually open the URL just given in the web browser yourself. In
RStudio, you may need to click ‘Open in Browser’ on top of the
Rstudio-window. The R session itself will produce copious amounts of
warnings which you can ignore. It can take up to 10 seconds or so to
become active; it may help to click a few of the buttons.
CHETAHshiny(ovarian.sce, redD=dim.red.u)
Explanation of the CHETAHshiny interface
The left column / margin shows parameters and thresholds that can be chosen. The top row shows which views there are. Many of the elements are clickable or provide info when you hover over them.
Classification tab
Shows the final classification and, further down, the statistics per cell type and the classification tree that was used (colors are consistent).
- you can zoom in, pan, hover, etc. Single-click a cell type adds/removes cells of that type from view. Double-click to show just that celltype or return to showing all cells
- ‘Color the intermediate types’ has the same effect as
PlotCHETAH(..., interm=TRUE, …) - The confidence score works as
Classify(..., thresh=some.value, ...)
Confidence scores tab:
The confidence score of cell in CHETAH represents the amount of evidence
that is available to continue classifying that cell further down the
classificatin tree. Choose a node (see the tree at the bottom) shows
only the cells classified to the specified node or more specific
(i.e. further down), and uses color to represent the confidence. The
fainter the color, the less evidence remains to continue making that
cell’s type more specific. The confidence score is, by definition,
positive, but here, negative values and colorscale are used to show
which of the two branches, if any, is to be taken by the cells in that
node. The colorcode is the same as shown in the tree under it. The
Profile score heatmap shows the corresponding profile scores (see below)
Raising the confidence threshold results in fewer cells ‘reaching’ the
selected node, so fewer cells will show up.
Profile score tab:
The profile score in CHETAH represents the similarity of cell in a particular node to any of the final types. If the confidence score (see above) is not below the threshold, the branch containing the cell type having the highest porfile score is taken. The classification tree is again shown at the bottoom.
Note that the profile score can be negative, but still he the highest in a particular node. E.g., the Macrophage score of many the macrophages in Node8 is negative, but mostly less so than the dendritics. If the confidence score still exceeds the threshold, the branch containing Macrophage would still be chosen.
This plot always shows all points, regardless of confidence threshold (and colors are also independent of this).
‘In a boxplot’ panel: unfortunately broken currently.
Genes used by CHETAH tab:
In each node and for each final type in that node, CHETAH uses the 200
genes that have the maximum absolute difference in expression between
that final type, and the average of all the types in the other branch to
calculate the profiles scores. If you set ‘# of genes’ in this plot to
200, the heat map will show you the mRNA counts in the input data, of
these 200 genes, for the selected node and cell type. If you select
fewer genes it will show (of these 200 genes) the most highly expressed
genes in the input (this is the default), or if Genes with max.
difference in the INPUT is unticked, the 200 genes most highly
expressed in the reference. Scale Matrix will normalize the genes for
better visualisation.
The tree is again shown at the bottom.
If you check e.g Node6 (the one that separates CD4 and CD8 cells), you
should appreciate that using marker genes (CD4 and CD8A
respectively; can you spot them?) is not going to work: the data is
noisy, expression is low and the differences in expression of the
classical canonical marker genes is miniscule. You need extra evidence
in the form of extra genes.
(More cells than strictly relevant are shown.)
Expression per gene tab: obvious
Session info
sessionInfo()
## R version 3.5.1 (2018-07-02)
## Platform: x86_64-apple-darwin16.7.0 (64-bit)
## Running under: macOS Sierra 10.12.6
##
## Matrix products: default
## BLAS: /Users/philip/local/Cellar/r/3.5.1/lib/R/lib/libRblas.dylib
## LAPACK: /Users/philip/local/Cellar/r/3.5.1/lib/R/lib/libRlapack.dylib
##
## locale:
## [1] C
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] cowplot_0.9.4 SingleR_0.2.2
## [3] Matrix_1.2-17 CHETAH_1.1.1
## [5] ggplot2_3.1.1 Seurat_3.0.1
## [7] SingleCellExperiment_1.4.1 SummarizedExperiment_1.12.0
## [9] DelayedArray_0.8.0 BiocParallel_1.16.6
## [11] matrixStats_0.54.0 Biobase_2.42.0
## [13] GenomicRanges_1.34.0 GenomeInfoDb_1.18.2
## [15] IRanges_2.16.0 S4Vectors_0.20.1
## [17] BiocGenerics_0.28.0
##
## loaded via a namespace (and not attached):
## [1] corrplot_0.84 plyr_1.8.4 igraph_1.2.4.1
## [4] lazyeval_0.2.2 GSEABase_1.44.0 splines_3.5.1
## [7] listenv_0.7.0 digest_0.6.18 foreach_1.4.4
## [10] htmltools_0.3.6 viridis_0.5.1 gdata_2.18.0
## [13] memoise_1.1.0 magrittr_1.5 doParallel_1.0.14
## [16] cluster_2.0.9 ROCR_1.0-7 limma_3.38.3
## [19] globals_0.12.4 annotate_1.60.1 doFuture_0.8.0
## [22] R.utils_2.8.0 colorspace_1.4-1 blob_1.1.1
## [25] ggrepel_0.8.1 xfun_0.6 dplyr_0.8.1
## [28] crayon_1.3.4 RCurl_1.95-4.12 jsonlite_1.6
## [31] graph_1.60.0 iterators_1.0.10 survival_2.44-1.1
## [34] zoo_1.8-5 ape_5.3 glue_1.3.1
## [37] gtable_0.3.0 zlibbioc_1.28.0 XVector_0.22.0
## [40] kernlab_0.9-27 future.apply_1.2.0 prabclus_2.2-7
## [43] DEoptimR_1.0-8 scales_1.0.0 pheatmap_1.0.12
## [46] mvtnorm_1.0-10 edgeR_3.24.3 DBI_1.0.0
## [49] bibtex_0.4.2 Rcpp_1.0.1 metap_1.1
## [52] viridisLite_0.3.0 xtable_1.8-4 reticulate_1.12
## [55] bit_1.1-14 rsvd_1.0.0 mclust_5.4.3
## [58] SDMTools_1.1-221.1 GSVA_1.30.0 tsne_0.1-3
## [61] htmlwidgets_1.3 httr_1.4.0 gplots_3.0.1.1
## [64] RColorBrewer_1.1-2 fpc_2.1-10 modeltools_0.2-22
## [67] ica_1.0-2 pkgconfig_2.0.2 XML_3.98-1.19
## [70] R.methodsS3_1.7.1 flexmix_2.3-15 nnet_7.3-12
## [73] locfit_1.5-9.1 labeling_0.3 tidyselect_0.2.5
## [76] rlang_0.3.4 reshape2_1.4.3 later_0.8.0
## [79] AnnotationDbi_1.44.0 pbmcapply_1.4.1 munsell_0.5.0
## [82] tools_3.5.1 RSQLite_2.1.1 ggridges_0.5.1
## [85] evaluate_0.13 stringr_1.4.0 yaml_2.2.0
## [88] bioDist_1.54.0 npsurv_0.4-0 outliers_0.14
## [91] knitr_1.22 bit64_0.9-7 fitdistrplus_1.0-14
## [94] robustbase_0.93-4 caTools_1.17.1.2 purrr_0.3.2
## [97] RANN_2.6.1 dendextend_1.10.0 pbapply_1.4-0
## [100] future_1.12.0 nlme_3.1-139 whisker_0.3-2
## [103] mime_0.6 R.oo_1.22.0 shinythemes_1.1.2
## [106] compiler_3.5.1 plotly_4.9.0 png_0.1-7
## [109] lsei_1.2-0 geneplotter_1.60.0 tibble_2.1.1
## [112] stringi_1.4.3 lattice_0.20-38 trimcluster_0.1-2.1
## [115] pillar_1.3.1 Rdpack_0.11-0 lmtest_0.9-37
## [118] data.table_1.12.2 bitops_1.0-6 irlba_2.3.3
## [121] gbRd_0.4-11 httpuv_1.5.1 R6_2.4.0
## [124] promises_1.0.1 KernSmooth_2.23-15 gridExtra_2.3
## [127] codetools_0.2-16 MASS_7.3-51.4 gtools_3.8.1
## [130] assertthat_0.2.1 withr_2.1.2 sctransform_0.2.0
## [133] GenomeInfoDbData_1.2.0 diptest_0.75-7 grid_3.5.1
## [136] tidyr_0.8.3 class_7.3-15 rmarkdown_1.12
## [139] Rtsne_0.15 singscore_1.2.2 shiny_1.3.2